 Login / Register
Login / Register 



Western blotting is a key tool in life science research that is used to separate and identify specific proteins from complex mixtures. We’ve developed a comprehensive selection of highly specific antibodies, ready-to-use buffers, and protein ladders. Try our reagents to obtain distinct and easy-to-interpret bands on your blots.
Loading...
Western Blotting
Western Blotting Introduction
Western blotting refers to a routine technique used to separate and identify proteins from complex mixtures. Proteins are separated (potentially under denaturing conditions) in a gel by size before being transferred to a membrane. The protein of interest is probed with specific antibodies and detected through a number of means, commonly chemiluminescence.
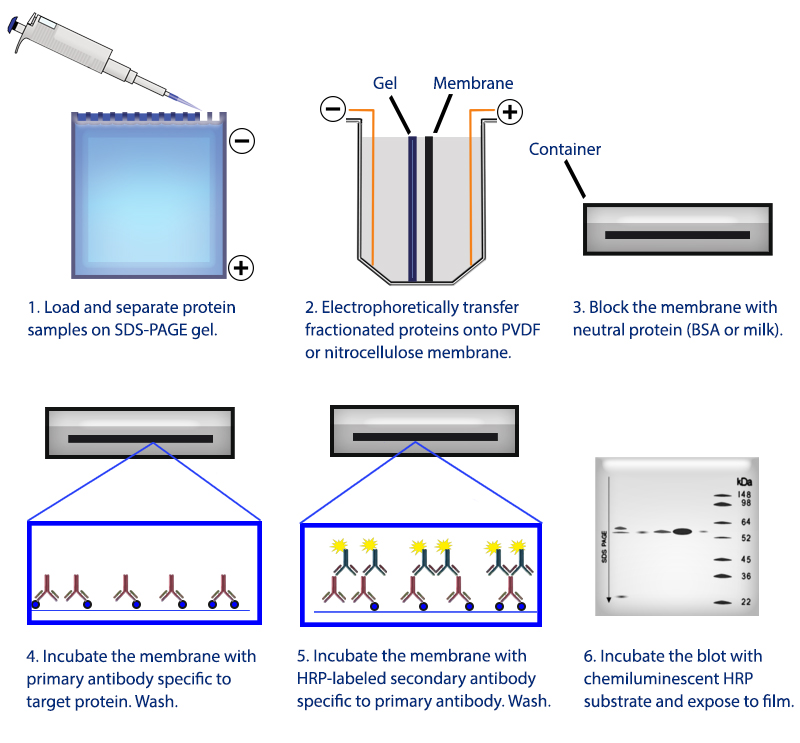
Figure 1. Overview of the western blotting procedure.
Sample Preparation Guide
For western blotting, sample preparation is a key step in the western blotting process. Proper sample treatment ensures you are able to detect proteins of interest. Below, you will find the general methodology used at BioLegend. You may wish to consult literature for more specific protocols relating to your samples of interest.
For cell culture samples
I. Isolation of cells: Isolate and wash the cells using ice-cold or pre-chilled PBS. For adherent cells, scrape the cells using a cell lifter. For suspension cells, centrifuge suspension to separate the cell pellet.
II. Cell lysis: Lyse the cells using a lysis buffer with added protease inhibitor.
III. Sonication: Sonicate the crude cell lysate to disrupt the highly viscous cellular DNA.
IV. Centrifugation: Centrifuge the sonicated lysate and collect the supernatant.
V. Protein estimation: Protein concentration is estimated using BCA or Bradford assay.
VI. Sample dilution and heat treatment: After protein estimation, dilute the samples with gel loading buffer/sample buffer and heat for 5-10 mins at 95-100°C.
For tissue samples
I. Homogenization: Smaller solid tissues are soaked in ice-cold lysis buffer and homogenized by using an electric homogenizer. For larger tissue samples, a blender is used to homogenize the tissue in PBS followed by treatment with lysis buffer to disrupt the cell membrane.
II. Centrifugation: Centrifuge the lysate and collect the supernatant.
III. Protein estimation: Protein concentration is estimated using BCA or Bradford assay.
IV. Sample dilution and heat treatment: After protein estimation, dilute the samples with gel loading buffer/sample buffer and heat for 5-10 mins at 95-100°C.
To start a western blotting procedure, gel electrophoresis is used to separate macromolecules in a sample. The most common type of electrophoresis used is called SDS-PAGE (SDS-Polyacrylamide Gel Electrophoresis). SDS is a type of detergent that adds a negative charge to amino acids in a protein, this along with heat applied during sample prep disrupts the tertiary and secondary structure of the protein. Because of the SDS, all proteins will have the same negative charge, resulting in separation being based on size rather than charge.
Once an electrical field is applied to the gel, small protein molecules move quickly the through the gel matrix toward the positive electrode, while larger proteins move through more slowly, resulting in a series of bands containing proteins of a particular size (Figure 2).
Protein samples are run along side a protein ladder containing several standards of known molecular weights. By using a ladder, the size of proteins in the sample lanes can easily be determined. BioLegend offers the Prime-Step™ Prestained Broad Range Protein Ladder, a three-color protein standard with 10 pre-stained chromophore-conjugated recombinant proteins covering a wide range of molecular weights, from 6.5 to 270 kDa.
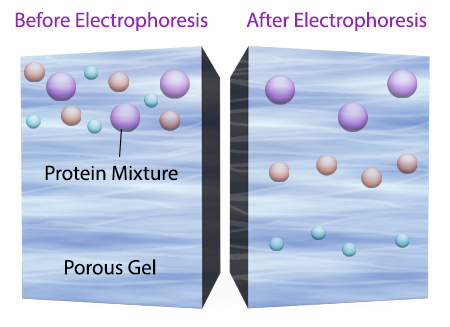
Figure 2. Negatively charged small protein molecules move through the gel matrix, toward the positive electrode, more quickly than larger negatively charged protein molecules.
After gel electrophoresis separated proteins on the gel are transferred onto a nitrocellulose or polyvinylidene difluoride (PVDF) membrane, again utilizing electrophoresis (Figure 3). The membrane is then blocked with neutral proteins, such as BSA or milk, to prevent non-specific binding of antibodies to the surface of the membrane.
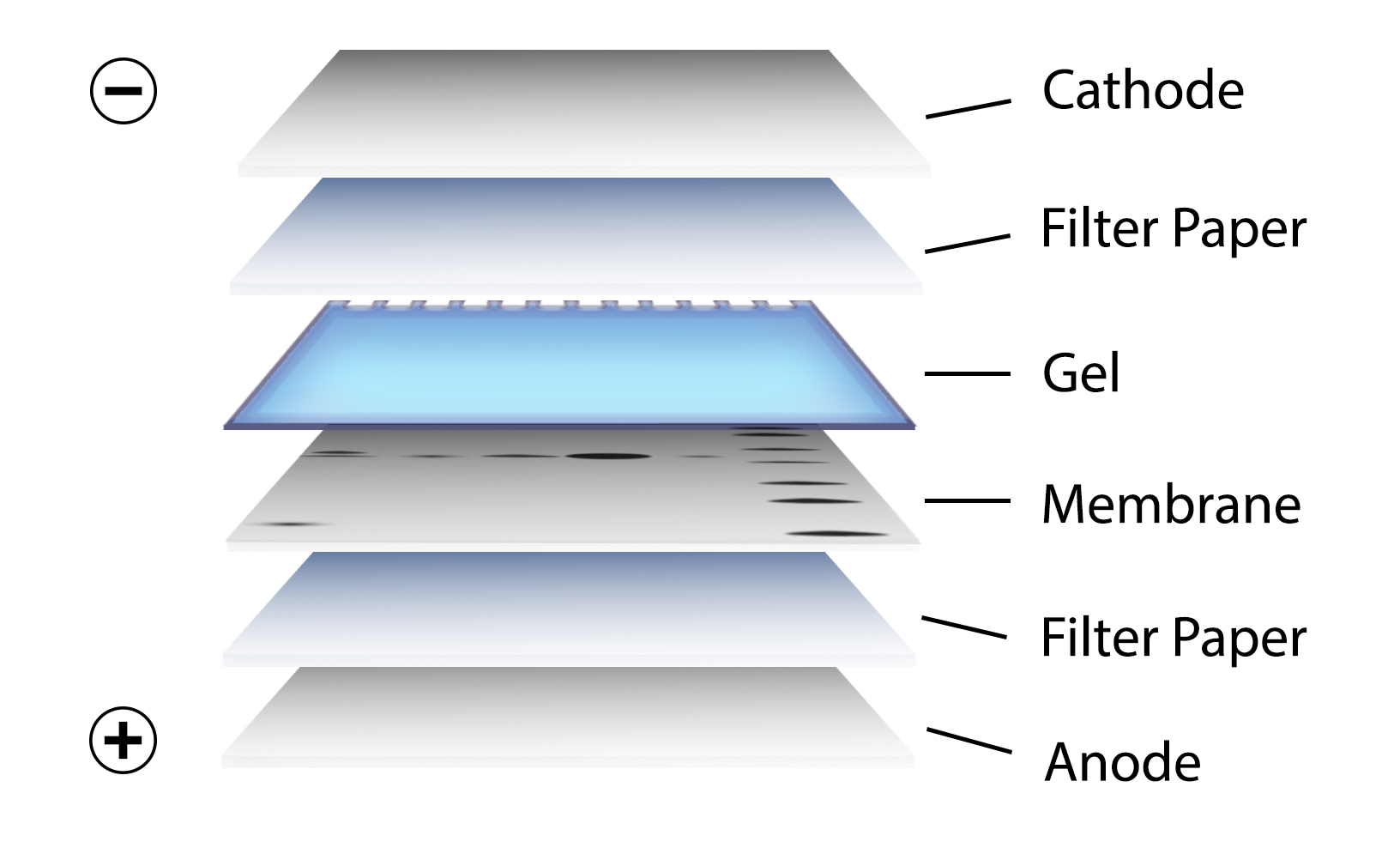
Figure 3. Transfer of the protein bands resulting from SDS-PAGE on to a membrane by electrophoresis
Antibody Binding/Target Detection
It is important to recall that SDS treatment of samples denatures proteins, causing them to lose their native conformation. This is why conformation epitope-specific antibodies and even flow cytometry antibodies may not always work in a western blotting assay. After transfer, the membrane is incubated with primary antibodies that bind specifically to the target protein, the primary antibody is not typically directly detectable. In colorimetric and chemiluminescent detection methods, the membrane is subsequently incubated with a detectable tagged secondary antibody specific to the host species of the primary antibody. Enzyme reporters like alkaline phosphatase (ALP) and horseradish peroxidase (HRP) are then used to generate signal in combination with a substrate like our Western-Ready™ ECL Substrate Kit. Direct-Blot™ products allow users to utilize primary antibodies directly conjugated to HRP, avoiding the need for a secondary reagent and streamlining the western blot process. For fluorescence-based detection methods, fluorophore-conjugated primary antibodies are used. These fluors typically emit in the near infrared range for detection.
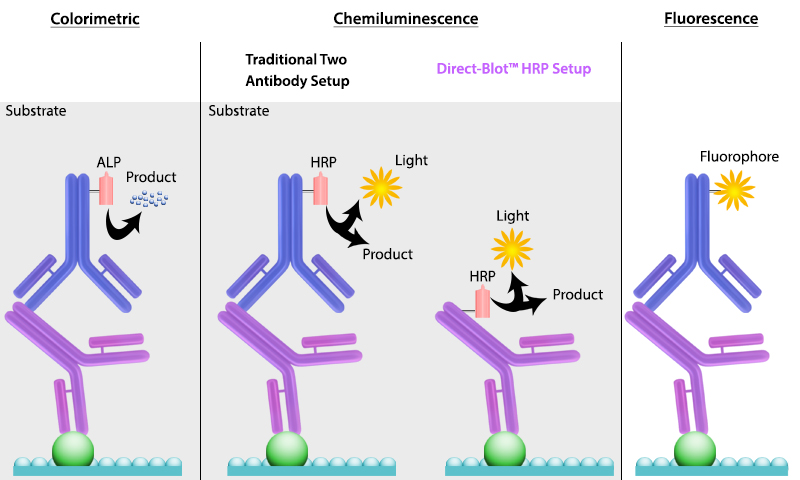
Figure 4. There are multiple ways to detect proteins of interest in a western blot. The colorimetric method detects signal via colored precipitate. In a chemluminescence-based method, light is emitted by the reaction shown. And finally, in fluorescence, a fluorophore-labeled antibody emits the signal.
Although, western blotting is a well-documented immunoassay, perfecting and optimizing your protocol can be tough. There are a number of common issues that may arise when developing new protocols for various reagents. Below you will find a comprehensive guide to help you troubleshoot common issues reported when western blotting.
You can also take a look at our recommended western blotting protocol for additional help. Or, contact tech@biolegend.com for more troubleshooting information.
Sections:
Multiple or Non-Specific Bands
Target Band is Extremely High/Low on Blot
| BioLegend Western Blot Troubleshooting Guide | ||
| What's the issue? | Possible Causes | Solution |
| No or low Signal | The primary and secondary antibodies are not compatible. | Ensure you are using secondary antibody that binds to your primary antibody (i.e. if your primary is rat, be sure you are using an anti rat secondary). |
| Insufficient primary or secondary antibody has bound to the protein of interest. | Utilize a higher concentration of antibody and or incubate for a longer period (i.e. overnight at 4°C). | |
| There is not enough antigen. | Load a larger amount of protein onto the gel. Use protease inhibitors and run the recommended positive control. | |
| Overuse of primary antibody. | Use fresh antibody (the effective concentration is lowered after each use). | |
| Incubation with detection reagent not sufficient. | Increase the blots incubation time with detection reagent. | |
| Detection reagents are not working. | Make sure detection reagents are functional by testing with a different primary antibody. | |
| Poor transfer during blotting. | Make sure the transfer apparatus is set up correctly. Ensure you are using the correct transfer times. | |
| Secondary antibody is inhibited by sodium azide. | Do not use sodium azide with HRP-Conjugated antibodies. | |
| Excessive membrane washing. | Reduce washing step repetitions or duration. | |
| Target protein ran off the gel. | Use a positive control and a molecular weight marker matched to the size range of the target protein. | |
| The target protein is not found in high concentrations in your sample. | Maximize the target's concentration by enriching the sample beforehand. | |
| The primary or secondary antibody binds to the blocking agent. | Utilize a mild detergent or switch to a different blocking reagent. | |
| Poor binding of proteins to membrane. | Use a membrane with the correct binding capacity. Dry PVDF membranes after transfer to promote strong binding. | |
| The protein in the species tested is not recognized by your primary antibody. | Run a positive control. Check literature or perform a BLAST alignment to see whether your antibody should react with the target protein. | |
| High Background | Insufficient washing or blocking. | Increase blocking time or consider using an alternate blocking reagent. Increase the number of washes. |
| Concentration of the primary antibody is too high. | Determine optimal antibody concentration through titration. Use a more dilute antibody with longer incubation times (slow targeted binding is best). | |
| Secondary antibody binding non-specifically, or binding with blocking agent. | Run a secondary control with no primary antibody. | |
| Overloaded protein. | Decrease the amount of protein loaded on the gel, or dilute the sample. | |
| Contamination of equipment or reagents. | Replace reagents, ensure all equipment is properly cleaned. | |
| Membrane causing high background. | PVDF membranes are considered to give higher background than nitrocellulose membranes. | |
| Membrane dried out during incubation. | Ensure membrane is not drying out during the incubation period. | |
| Incubation temp too high. | Incubate membrane at 4°C. | |
| Cross-reactivity of phospho-specific antibodies with blocking agent. | The user may have to try different blocking buffers, such as milk, BSA, etc. to reduce non-specific binding while maintaining specific signals. For optimal results, follow the blocking buffer recommendations from your antibody provider. | |
| Multiple or non-specific bands | Primary antibody concentration too high. | Decrease the concentration of primary antibody. Run secondary control without the primary antibody. |
| Excess protein on gel. | Reduce the amount of protein loaded. | |
| Issues with blocking. | Optimize blocking time and blocking reagent. | |
| Insufficient washing. | Increase number of wash steps. | |
| Antibody not properly purified. | Use antibodies purified by the affinity method. | |
| Target protein has been degraded. | Use fresh sample. Include protease inhibitors in your sample buffer. | |
| Frequently passaged cell lines accumulate differences in protein expression profiles. | Retrieve and expand original cell line, run samples in parallel. | |
| Target protein has several modified forms (acetylation, methylation, glycosylation etc.). | Refer to literature, use agent to remove modifications when possible/necessary. | |
| Protein subtypes have different molecular weights. | Use bioinformatics analysis and review literature to estimate the correct protein size. | |
| There are splice variants from the same protein family that share similar epitopes. | Check literature and/or perform a blast search to confirm. | |
| Multimer formation of target protein. | Prior to SDS page, boil protein for 10 min to disrupt multimers. | |
| Diffuse Bands | Concentration of antibody too high. | Reduce antibody concentration. |
| Protein transfer too rapid or gel became over-heated during electrophoresis. | Increase the transfer time and/or run gel at 4°C | |
| Too much protein loaded on gel. | Decrease the quantity of protein loaded on gel. | |
| Smile effect on bands | Migration was too rapid. | Decrease voltage when running gel. |
| Temperature during migration was too high. | Run gel at 4°C. | |
| Uneven staining of gel | Bacterial contamination of antibodies. | Store antibodies as recommended by your antibody provider. Use fresh buffers. |
| Insufficient antibody volume. | Ensure that the membrane is completely covered with antibody and incubate with agitation. | |
| Target band is extremely high/low on blot | Separation during electrophoresis not efficient. | Change gel percentage: use a lower percentage for large proteins, a higher percentage for small proteins. |
| Lane with protein ladder is black | The antibody is reacting with the protein ladder. | Load gel so there is a blank lane between the ladder and the first sample lane. |
| Uneven white spots on blot | Air bubbles were trapped between the gel and membrane during transfer. | Ensure air bubbles are removed when preparing for transfer. |
| Black dots on the blot | Antibodies are binding to blocking agent. | Filter blocking agent. |
When performing a western blot, secondary antibodies are commonly used to detect your protein of interest. However, unless the signal needs to be amplified, there is no advantage to using a two-step process. Our Direct-Blot™ reagents are horseradish peroxidase (HRP)-conjugated primary antibodies that eliminate the need for a secondary antibody. By removing several steps from the workflow, they shorten the protocol and help you get your results faster. Our scientists have carefully developed and tested these antibodies to ensure they provide high sensitivity and excellent stability.
Follow our Direct-Blot™ protocol and learn more about Direct-Blot™ antibodies.
Direct-Blot™ HRP anti-β-Actin Antibody
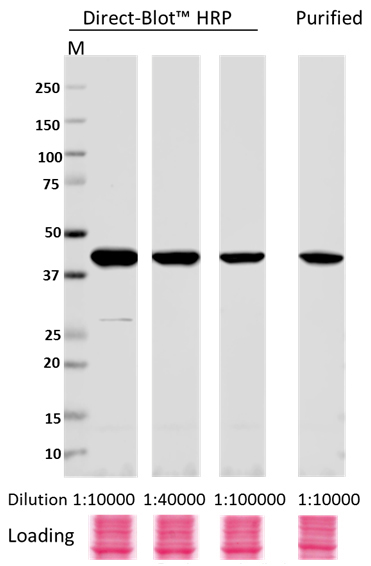
Whole cell lysates (15 µg protein) from HeLa cultures were resolved by electrophoresis (4-20% Trisglycine gel), transferred to nitrocellulose, and probed with 1:10,000, 1:40,000, and 1:100,000 dilutions of Direct-Blot™ HRP anti-β-actin Antibody, clone W16197A, or a 1:10,000 dilution of purified anti-β-actin Antibody, clone W16197A (upper). Proteins were visualized using a 1:3000 diluted HRP anti rat-IgG secondary antibody for purifi ed anti-β-actin Antibody and chemiluminescence detection. Ponceau S staining was used as loading control (lower). Lane M: MW ladder.
Antibodies
Primary Antibodies
To detect your protein of interest, we provide highly specific primary antibodies validated and quality-tested by western blotting. Our antibodies cover a wide range of targets across cell biology, neuroscience, immunology, and other key signaling pathways. These come in either a purified, unconjugated format or directly conjugated to HRP.
Find Western Blotting Primary Antibodies
Secondary Reagents
Secondary reagents target the primary antibody and are conjugated to provide visualization of the protein of interest. For western blotting, these reagents are conjugated to HRP which is used as a reporter enzyme.
Knockout/Knockdown Validation
As knocking out the target protein is one of the most trusted antibody validation processes, we validate many of our cell biology antibodies by KO (knockout) and KD (knockdown) systems.
The reproducibility of published research has emerged as an urgent topic in today’s scientific community. From funding agencies to researchers to manufacturers and publishers, it is critical for all of these groups to align themselves to ensure that research is done with rigor and is reproducible.
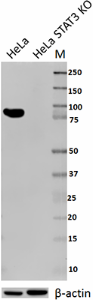
Total lysates from HeLa and HeLa STAT3 CRISPR/Cas9 knockout (KO) cells were probed with 1 µg/ml purified anti-STAT3 antibody. Proteins were visualized using chemiluminescence detection by incubation with HRP goat anti-mouse-IgG secondary antibody. Direct-Blot™ HRP anti-β-actin Antibody was used as a loading control (lower).
PhosphoPair Antibody Set
Try PhosphoPair Antibody Sets for a convenient, cost-saving solution to interrogate phosphorylation and cell signaling pathways. Each set bundles an antibody specific for a phosphorylated protein with one that recognizes total protein. Sets are available for critical signaling mediators like STAT3, ERK, and mTOR.
Key Features:
- Each antibody is quality-tested by western blot.
- Supplied in a purified format at 25 µg per vial.
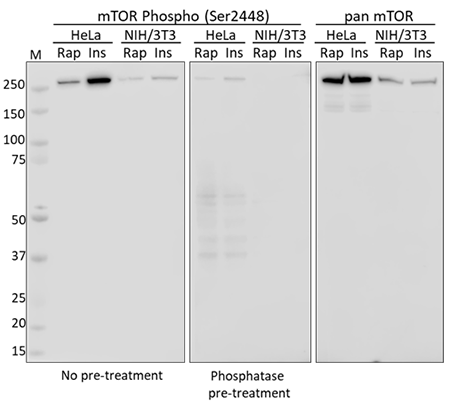
Western blot analysis using whole cell extracts from serum starved HeLa and NIH/3T3 cells treated with rapamycin (Rap) or insulin (Ins). Membranes were probed with purified anti-mTOR Phospho (Ser2448) and a duplicate membrane was pre-treated with lambda protein phosphatase prior to incubation. Equal mTOR loading was confirmed by probing membranes with an anti-pan mTOR antibody.
Antibody Sampler Kits
Antibody sampler kits provide a convenient and economical resource to test multiple antibodies. Each kit contains at least 4 antibodies kits and target cell function and structure proteins, related detection methods (such as epitope tags), or proteins connected through signaling pathways.
Protein Ladders
Our Prime-Step™ Prestained Broad Range Protein Ladder is a three-color protein standard with 10 prestained chromophore-conjugated recombinant proteins covering a wide range of molecular weights—from 6.5 to 270 kDa. It is ideal for monitoring protein separation during SDS-Page, confirming WB transfer on membranes, and estimating protein sizes.
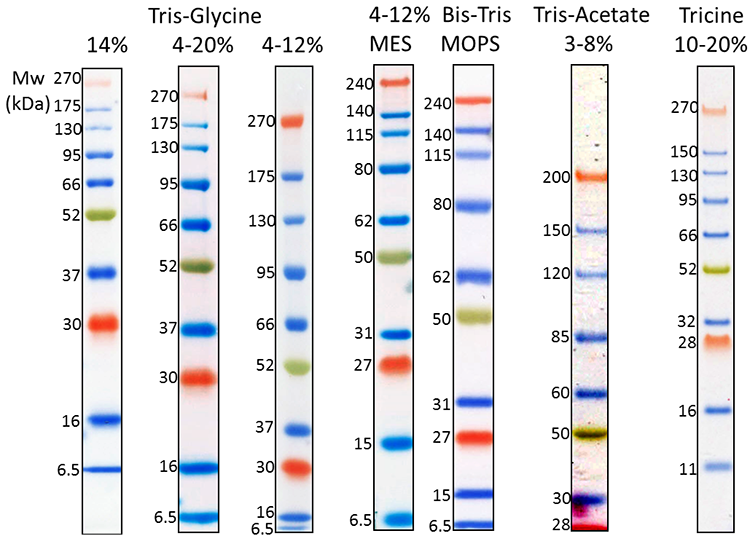
3 µL Prime-Step™ Prestained Broad Range Protein Ladder was resolved by electrophoresis in different gel running systems, including 14%, 4-20% and 4-12% Tris-Glycine gel, 4-12% Bis-Tris gel (MOPS and MES running buffer), 3-8% Tris-Acetate gel and 10-20% Tricine gel. Protein ladders were transferred to a nitrocellulose membrane.
ECL Substrates
Our Western-Ready™ ECL Substrate Kits are a non-radioactive enhanced luminol-based chemiluminescent substrate (ECL) for the detection of horseradish peroxidase (HRP) activity from WB reagents and ELISA immunoassays. Our kits provide sensitive detection and sustained signal for up to 4 hours. Our ECL Plus Kit is ideal for detecting low- to mid- picogram levels of antigen, while the Premium Kit provides more sensitive detection of high-femtogram levels of protein.
Key features:
- Luminol-based detection system
- Sensitive detection
- Available in small 20 mL size for sampling
- Multiple kits with a range of sensitivities
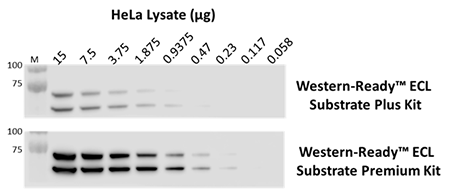
The indicated amounts of whole cell extract from HeLa cells were probed with purified anti-Lamin A/C antibody. Proteins were visualized using HRP goat anti-mouse IgG antibody, followed by incubation with Western-Ready™ ECL Substrate Plus Kit or Western-Ready™ ECL Substrate Premium Kit.
Western-Ready™ Buffers
Try our complete set of Western-Ready™ buffers for every step of your workflow, including protein extraction, gel loading and running, and detection.
|
Description |
Use |
|
Running buffer for resolving small-to-medium-sized proteins on Bis-Tris SDS page gels. |
|
|
Used for the preparation of samples for non-reducing and reducing SDS-PAGE. |
|
|
Stringent lysis buffer to extract proteins from all subcellular compartments. |
|
|
Buffer for blocking, washing, and antibody dilution. Formulated to reduce non-specific binding. |
|
| Western-Ready™ Transfer Buffer (10X) | Buffer formulated for western blot protein transfer onto PVDF or nitrocellulose membranes. |
ProductsHere



Follow Us