 Login/Register
Login/Register 



Single-Cell Multiomics: A Beginner’s Guide to Understanding CITE-Seq and TotalSeq™ Reagents
Single-cell multiomics is a burgeoning field that integrates multiple omics research areas, like transcriptomics and proteomics, and generates large datasets that reveal new insights into health and disease. As the field rapidly evolves, it can be difficult to keep track of the terminology and applications. In this blog, we’ll review the basics of proteogenomics, how it has expanded, and importantly, how our TotalSeq™ reagents enable these applications.
What are CITE-Seq and REAP-seq?
Single-cell isolation and RNA sequencing introduced a leap forward for biology by improving analytical resolution to the single-cell level. By analyzing transcriptional changes in individual cells, bioinformatics has identified new populations of cells and previously unseen differences between experimental and control conditions.
Flow cytometry has been a popular technique employed for decades to characterize and analyze cell populations using cell surface markers. However, one major limitation of flow cytometry is that the number of markers that can be used to simultaneously identify cell populations is dependent on the spectral overlap of fluorophores. As flow cytometry-based techniques continue to advance, the number of markers grows: 10-20 markers using standard flow cytometry; 30+ markers using spectral cytometry; and 40-50 using CyTOF®.
Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq), developed by the New York Genome Center (NYGC), brings together surface protein phenotyping (similar to flow cytometry) and single-cell RNA sequencing (scRNA-seq)1. This is achieved by using antibodies conjugated to oligonucleotides that can be captured in a similar way to mRNA on a scRNA-seq platform. A similar technique was also published around the same time called RNA Expression and Protein Sequencing assay (REAP-seq)2. Since these techniques use oligonucleotides instead of fluorophores and rely on sequencing as a readout, there is no known limit for the number of simultaneous markers that can be analyzed at one time. We currently offer a TotalSeq™-A Human Universal Cocktail v1.0 that contains 163 antibodies, and our scientists have utilized 200+ markers for internal and custom applications.
How Does it Work?
Typical high-throughput single-cell sequencing platforms take advantage of beads covered in poly(dT) oligonucleotides that capture mRNA by hybridizing with their poly(A) tail. These oligonucleotides on the bead also contain additional sequences. The cell barcode is used to associate the sequence to a specific cell and a unique molecular identifier (UMI) distinguishes the specific mRNA molecule.
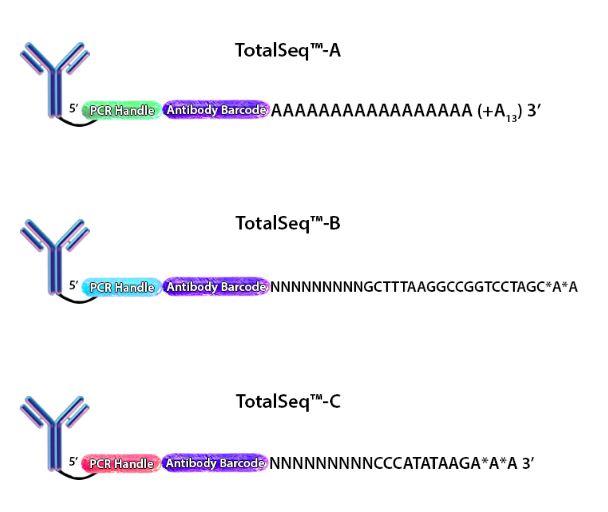
Figure 1. Depiction of the oligonucleotides conjugated to each of our TotalSeq™ antibodies.
When cells stained by these antibodies are lysed, the mRNA is captured by the bead and the ADT is also captured in an identical fashion. The captured mRNA can be processed into a scRNA-seq library. Since the ADTs are short, they can be isolated from the cDNA by size using magnetic beads. ADTs are then processed into an independent ADT library because they have an incorporated PCR handle. The ADT libraries can be sequenced independently, but they are usually pooled with their respective cDNA library. Typically, the ADT library represents 5-10% of the pooled libraries and is sequenced at a read depth of 5,000-10,000 reads/cell (depending on how many ADTs are used). Our TotalSeq™-A line of reagents was designed with a poly(A) sequence, making it platform agnostic and allowing it to work with most, if not all, poly(dT) capture methods.
Newer Iterations of CITE-seq
Since the development of CITE-seq and REAP-seq, TotalSeq™ antibodies have been used in an expanding number of applications and with new single-cell platforms.
TotalSeq™-B utilizes 10x Genomics’ Feature Barcoding technology, which has been implemented on the 10x Genomics’ 3’ Gene Expression v3+ kits. Feature barcoding technology is an independent capture sequence that is on the surface of the beads. Currently, there are two capture sequences, 1 and 2. These capture sequences were added to the beads to support the exclusive capture of other designed reagents, including oligonucleotide tags on antibodies or single-guide RNA (sgNRA) from CRISPR screening, independent of mRNA capture by poly(dT). TotalSeq™-B has been designed to be captured by one of these sequences.
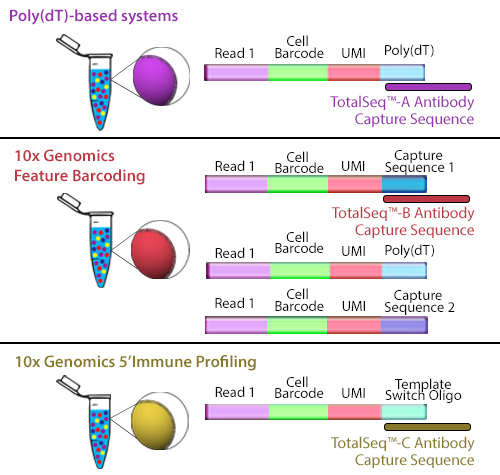
Figure 2. Illustration depicting how each TotalSeq™ antibody format binds to capture beads.
Though they use different capture sequences, there are no significant differences in identifying positive or negative populations when using either poly(dT) capture with TotalSeq™-A or feature barcode capture using TotalSeq™-B reagents. While there may be a slightly lower capture of mRNA when using TotalSeq™-A since there is competition for binding sites, this difference is negligible. The rate of capture between TotalSeq™-A and -B are inherently different, but since the libraries are processed independently, they are comparable bioinformatically. While TotalSeq™-A and B can be used simultaneously, it can be difficult to bioinformatically deconvolute the data, and therefore, is not advised.
TotalSeq™-C functions similarly to TotalSeq™-A; however, TotalSeq™-C is designed to work with 10x Genomics’ 5’ Immune Profiling kits. Since this platform utilizes a 5’ capture, the beads do not contain a poly(dT) capture sequence. Instead, an anti-template switching oligo (TSO) sequence is used. TotalSeq™-C antibodies contain an oligonucleotide sequence that has been altered to suit this different capture method.
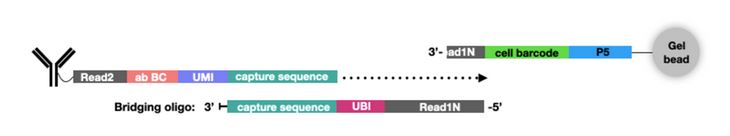
In addition to new single-cell platforms, CITE-seq has been expanded to include additional multiomics parameters. For example, ATAC with Select Antigen Profiling by sequencing (ASAP-seq) brings CITE-seq technology to single-cell ATAC sequencing (scATAC-seq) to assess genome-wide chromatin accessibility. scATAC-seq beads have a unique capture sequence from those discussed above. These capture sequences have a cell barcode but no UMI, since there are only two copies of a gene in a cell. Instead of designing new oligo tags for the new capture sequence like TotalSeq™-C, a bridging oligo is used (Fig. 3). This bridging oligo is able to bind to the capture sequence on the bead as well as the capture sequence of the ADT. The bridging oligo also contains a unique bridging identifier (UBI) to add the resolution and controlling for PCR duplication similar to a UMI.
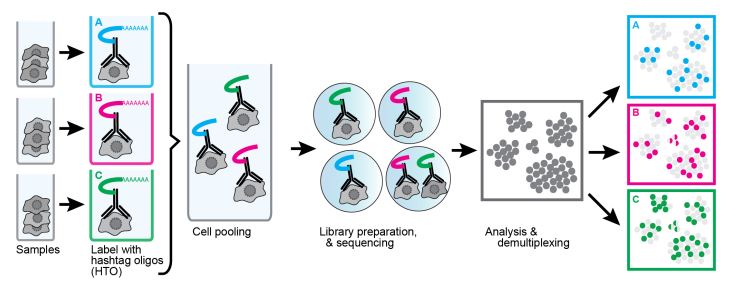
Cell Hashing
Another useful technique using oligonucleotide-conjugated antibodies is cell hashing. Using antibodies against markers that are ubiquitously expressed across different cell types, each sample can be tagged with a different oligonucleotide barcode. Our TotalSeq™-A, -B, and -C formats contain hashtag antibodies available for human and mouse specificities. TotalSeq™ hashtag reagents are a mix of two clones. Human hashtag reagents target CD298 and β2 microglobulin, and mouse hashtag reagents bind CD45 and MHC class I (a, b, d, j, k, s, and u haplotypes). This allows for the tagging of separate samples, which can then be pooled into an independent single-cell run (Fig. 4).
The labeled cells are identified by their unique hashtag and assigned to their sample group. This has the added benefit of identification of doublets/mutiplets. If a data point has more than one hashtag, it is very likely to be two or more cells that were captured together. Unfortunately, this doesn’t account for cells with the same hashtag being captured together. Still, being able to identify mutiplets opens the possibility of “super loading” and expanding the number of loaded cells. Without hashtagging, this is difficult as an increase in input cells in a single-cell sequencing experiment increases the likelihood of doublet formation. This limits the number of cells that can be loaded, as doublets/multiplets would begin to occupy too many data points. By hashtagging samples and identifying multiplets for exclusion, many more cells can be super loaded compared to conventional methods. It should be noted that super loading does have diminishing returns as the doublet rate increases.
Nuclear hashtags are also available in our TotalSeq™-A format. These bind to nuclear pore complex proteins and retain all of the cell hashing benefits for nuclei in single-nucleus RNA-seq (snRNA-seq) experiments.
BEN-seq
TotalSeq™ antibodies are not only useful in single-cell sequencing experiments, but also in bulk RNA-seq. In collaboration with Illumina, we developed a Bulk Epitope and Nucleic acid sequencing (BEN-seq) workflow to leverage the ability to measure surface proteins by sequencing. Samples are processed in a similar way to CITE-seq, except that the readout provides a holistic view of surface protein expression and cDNA from a population of cells. BEN-seq can easily be adapted to work with typical bulk RNA-seq workflows and reagents. Learn out more about BEN-seq in our application note.
Multiomics has transformed traditional sequencing experiments and can provide unparalleled protein and genetic analysis in a single experiment. Explore our TotalSeq™ reagents and see how they can be incorporated into your workflow. And if you can’t find the reagents you’re looking for, contact us to discuss custom oligo-conjugated antibodies, hashtags, or antibody cocktails for your experiment.
References:
- Stoeckius et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017 Sep; 14(9): 865-868.
- Peterson et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017 Oct; 35(10): 936-939.



Follow Us