 Login/Register
Login/Register 



Parkinson's disease is one of the most well-known neurodegenerative disorders among the 600+ disorders afflicting the nervous system. This page details the known associated proteins that contribute to the disease. BioLegend provides an array of world-class antibodies, ELISA kits, and recombinant proteins for Parkinson’s research.
Loading...
Parkinson's Disease
Background
Parkinson's disease (PD) is the second most common neurodegenerative disorder after Alzheimer's disease (AD), affecting 1–2% of the population over age 60. PD is characterized by the progressive degeneration and loss of dopaminergic (DA) neurons in the substantia nigra of the midbrain, and defined in part by the presence of intracytoplasmic inclusions called Lewy bodies (LBs). PD manifests as a variety motor symptoms such as resting tremors, rigidity and postural instability. PD is often accompanied by non-motor symptoms (NMS), which may occur in the pre-motor phase of the disease and include olfactory dysfunction, REM sleep disorder, anxiety and depression. Cognitive impairment affecting executive functions such as attention, recognition, working memory, and problem solving may also appear in the pre-motor phase and progressively increase in intensity leading to dementia in the late phase of the disease.

Sporadic forms of PD constitute the majority of affected population and patients are generally afflicted later in life, with the onset of disease occurring over the age of 60. Individuals with genetically-linked familial forms of PD, represent less than 10% of the patients, and have earlier onset of disease; occurring before the age of 40 or 50. Mutations in the genes that code for α-synuclein, Parkin, leucine-rich repeat kinase 2 (LRRK2), PTEN-induced putative kinase 1 (PINK1), and protein deglycase DJ-1 have been associated with developing PD. Currently, there are no laboratory tests to help diagnose sporadic Parkinson's disease. A definite diagnosis is only accomplished based on post-mortem immunohistochemical analysis and detection of specific inclusions (LBs) in the brain of affected individuals.
| Gene | Protein | Age of Onset | Inheritance | Products |
|---|---|---|---|---|
| PARK1/PARK4 | α-synuclein | Mid 20-30 | Dominant | View Products |
| PARK2 | Parkin | Juvenile to 40 | Recessive | View Products |
| PARK5 | UCH-L1 | 30-50 | Dominant | |
| PARK6 | PINK1 | 30-50 | Recessive | View Products |
| PARK7 | DJ-1 | 20-40 | Recessive | View Products |
| PARK8 | LRRK2 | 40-60 | Dominant | View Products |
α-Synuclein
α-synuclein is a small 140 amino acid (aa) protein that is abundantly expressed in the brain. α-synuclein is mainly localized at the pre-synaptic terminals where it is thought to play a role in regulating dopamine release by maintaining a supply of synaptic vesicles in the pre-synapse. A compelling body of work has provided a strong link between α-synuclein and PD. An imbalance between the rate of protein expression, clearance and aggregation leads to accumulation and aggregation of α-synuclein. Gene duplications and triplications affect α-synuclein synthesis and lead to increased intracellular levels of the protein. Oxidative stress, post-translational modifications and certain mutations in the α-synuclein gene have been shown to enhance its propensity to aggregate and accumulate. Failure to degrade α-synuclein caused by deficits in the protein clearance machinery, the Ubiquitin Proteasome System (UPS) and the autophagy pathway, also contribute to its buildup within a cell. Conversely, oligomeric or aggregated forms of α-synuclein may interfere with or disrupt the function of the cellular clearance machinery, and further block degradation of the accumulated protein. These factors lead to abnormal intracellular levels of α-synuclein, favor the formation/accumulation of toxic oligomeric and fibrillar species, and deposition into what eventually becomes a Lewy body. In fact, α-synuclein is the major component of LBs which are a pathological hallmark of several neurodegenerative disorders including Parkinson's disease, and are thought to be associated with neuronal cell dysfunction and death.

Proposed pathways involved in the aggregation of α-synuclein. Under normal physiological conditions, α-synuclein exists as a monomeric, natively unfolded protein. Monomeric α-synuclein is highly soluble and capable of binding to a variety of cellular membranes where it assumes an α-helical confirmation. Under pathophysiological conditions or at high concentrations, unfolded monomers undergo a conformational change to assume β-sheet-like structures which can self-associate to form small oligomeric species (e.g. dimers), high molecular weight insoluble fibrils, and trans-membrane pore-like structures composed of ring-like cytosolic oligomers. The accumulation of the fibrils leads to the formation of Lewy bodies. The aggregated and fibrillar forms of α-synuclein are known to be highly toxic, interfering with cellular functions such as mitochondrial function, endoplasmic reticulum–Golgi trafficking, protein degradation and synaptic transmission. Dysfunction in these pathway can ultimately lead to neurodegeneration. Furthermore, the ring-like pores can damage membrane integrity and disturb intracellular calcium homeostasis, contributing to neuronal toxicity

DJ-1
Protein deglycase DJ-1, commonly known as DJ-1, is a ubiquitously expressed protein that is localized to the cytoplasm, mitochondria and nucleus. DJ-1 is a multi-functional protein involved in cellular processes such as transcriptional regulation, oxidative stress response, and mitochondrial function. DJ-1 was originally identified as an oncogene but has since been implicated in other diseases such as ischemic injury and amyotrophic lateral sclerosis (ALS). It's association with Parkinson's disease arises from mutations and truncation in the protein, all of which lead to early-onset familial form of PD.

DJ-1's activity is regulated by its oxidative status. Oxidation of the cysteine residue C106 in DJ-1 contributes to its quenching activity against reactive oxygen species (ROS) produced under oxidative stress conditions. Thus, DJ-1 acts as a sensor of cellular redox metabolism and a scavenger of free radicals. Furthermore, oxidative stress leads to translocation of DJ-1 from the cytoplasm to the nucleus where it induces activation of anti-oxidant genes to reduce the levels of ROS. C106 is a highly conserved residue among species and important for all of the functions of DJ-1. The current model for DJ-1's involvement in the sporadic cases of PD proposes the up-regulation of its expression in response to oxidative stress. Under continuous oxidative stress, a known contributing factor to dopaminergic neuronal loss in PD, DJ-1 becomes highly oxidized leading to loss of its activity. Consistent with this model, aberrant oxidation of C106 has been observed in PD brain tissues. In the case of inherited forms of PD, mutations in the DJ-1 gene render it inactive and incapable of exerting its neuroprotective effect.

C106 is activated under oxidative stress conditions through sequential oxidation with SOH and SO2H. Excessive oxidation with SO3H is thought to render DJ-1 inactive.
Adapted from Ariga H et al., Oxidative Medicine & Cellular Longevity 2013
Of note, DJ-1 was shown to positively regulate tyrosine hydroxylase (TH) gene expression, and enhance TH activity under oxidative stress. TH is the enzyme responsible for converting the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA), the precursor to the neurotransmitter dopamine. Under normal conditions, dopamine is produced in the cytoplasm and immediately sequestered into vesicles. If unstored, dopamine can auto-oxidize and produce toxic molecules such as ROS. Interestingly, DJ-1 also upregulates the gene expression and activity of vesicular monoamine transporter-2 (VMAT2), a protein required for the uptake of excess dopamine into synaptic vesicles to prevent oxidized dopamine-induced damage in neurons. In PD, loss of DJ-1 function may render dopaminergic neurons unable to efficiently sequester dopamine into vesicles, leading to elevated oxidation of dopamine and increased production of toxic byproducts including ROS. Reactive oxygen species cause functional alterations in proteins, DNA and lipids. These oxidative insults, in turn, interfere with cellular processes which ultimately promote neurotoxicity.

Dopamine metabolism and release from dopaminergic neurons. Tyrosine is converted to L-DOPA and dopamine through sequential action of TH and DOPA decarboxylase, respectively. VMAT2 then mediates the translocation and sequestration of dopamine into DA vesicles. The release and uptake of dopamine by its receptors on the postsynaptic membrane leads to activation of signaling pathways involved in normal neuronal function. Excess dopamine is recycled back into the presynapse via dopamine transporter, or catabolized by the monoamine oxidase (MAO) and cathecol-O-methyl transferase (COMPT) enzymes. Inhibition of DJ-1 activity either through mutations or excessive oxidation may lead to repression of TH and VMAT2 expression, and elevated levels of cytosolic dopamine and dopamine-induced cytotoxicity.
Adapted from cummings JL et al., Brain 2011
PINK1
PTEN-induced putative kinase 1 (PINK1) is a serine/threonine protein kinase that is commonly mutated in familial forms of PD known as
autosomal-recessive juvenile Parkinsonism (AR-JP). PINK1 contains an N-terminal mitochondrial localization signal which allows its associated with the outer mitochondrial membrane where it interacts with its binding partners. PINK1 is responsible for surveying the mitochondrial health status by detecting and targeting damaged mitochondria for degradation.
Under healthy conditions where mitochondrial membrane potential is maintained, PINK1 is imported into the inner mitochondrial membrane (IMM) through interactions with mitochondrial translocase complexes, TOM and TIM. At the IMM, PINK1 is sequentially cleaved by MPP and PARL to remove its mitochondrial localization signal, and expose a region of PINK1 that signals it's targeting and degradation by the proteasome. This constitutive import and proteolysis of PINK1 ensures the maintenance of healthy mitochondria to meet the cellular energy demands. In damaged mitochondria, the loss of transmembrane potential leads to failure in transporting PINK1 to the IMM, and its accumulation at the outer mitochondrial membrane (OMM) where it remains bound to the TOM complex. Localization of PINK1 to OMM spares it from cleavage and subsequent proteasomal degradation. Instead, PINK1 undergoes autophosphorylation, and induces Parkin ligase activity and recruitment to the mitochondria. These proteins then initiate a cascade that leads to the degradation of damaged mitochondria through mitophagy. PINK1/Parkin-mediated mitophagy ensures the removal of toxic mitochondrial products and prevents neuronal loss. In PD patients, mutations in the PINK1 protein lead to accumulation of misfolded proteins in the mitochondria as well as fail to protect against stress-induced mitochondrial dysfunction.

Schematic representation and post-translational processing of PINK1. PINK1 is synthesized as a 63 kD protein and is targeted to the IMM via TOM/TIM complex. Full-length PINK1 is then processed by the mitochondrial processing protease (MPP) to remove its mitochondrial targeting sequence (MTS) and generate a 60 kD form. This MPP-cleaved form of PINK1 is further processed within the transmembrane domain (TMD) by PARL to produce a 52 kD form, which is released to the cytosol and degraded through the ubiquitin proteasome pathway.
Parkin
 Mutations in the Parkin gene were the first mutations found to be associated with autosomal-recessive juvenile PD. Parkin belongs to a family of E3 ligases responsible for transferring ubiquitin (Ub) moieties to substrates that are destined for degradation by the Ubiquitin Protease System (UPS). The function of Parkin has been linked to mitochondrial quality control and deemed to be necessary for mitophagy (autophagy of mitochondria).
Mutations in the Parkin gene were the first mutations found to be associated with autosomal-recessive juvenile PD. Parkin belongs to a family of E3 ligases responsible for transferring ubiquitin (Ub) moieties to substrates that are destined for degradation by the Ubiquitin Protease System (UPS). The function of Parkin has been linked to mitochondrial quality control and deemed to be necessary for mitophagy (autophagy of mitochondria).

Schematic diagram of Parkin and its domains. Parkin contains an N-terminal ubiquitin-like (UBL) domain, an atypical RING (really interesting new gene) domain (RING0), and a C-terminal domain consisting of two RING finger motifs (RING1 and RING2) that are separated by an in-between-RING (IBR) domain. Parkin is categorized as a HECT-RING hybrid E3 ligase meaning it can form an intermediate between ubiquitin and a catalytic cysteine residue (HECT-type E3 ligase) before transferring ubiquitin to a substrate. Parkin also exhibits RING finger-type E3 ligase activity by directly mediating the transfer of ubiquitin to its substrate. The UBL domain of Parkin inhibits its ligase activity by interacting with the IBR domain. Phosphorylation of S65 in the UBL domain and transient interaction with pS65-ubiquitin lead to a conformational change in Parkin and activation of its ligase activity.
Mitochondrial dysfunction is one of the major contributing factors in the pathogenesis of neurodegenerative disorders such as Parkinson's disease. PINK1 and Parkin work together in maintaining mitochondrial health, detecting damaged mitochondria and initiating cascades that lead to their degradation through mitophagy. In the cytosol, Parkin remains inactive by assuming an autoinhibited conformation. In response to mitochondrial damage, PINK1 initiates a cascade where it phosphorylates ubiquitin linked to mitochondrial proteins at S65 (pS65-Ub). Phosphorylation of ubiquitin drives the recruitment and translocation of cytosolic Parkin to the OMM, where binding to pS65-Ub destabilizes Parkin's autoinhibitory conformation, and promotes its ligase activity and ability to ubiquitinate OMM substrates. In this state, Parkin is phosphorylated by PINK1 which further stabilizes its active conformation and binding to pS65-Ub. This cascade serves as rapid response for translocation and activation of Parkin to induce mitophagy of damaged mitochondria. Activation of PINK1 and Parkin in response to mitochondrial damage leads to the coating of mitochondria with ubiquitinated mitochondrial proteins. These ubiquitin chains serve as a signal for the recruitment of the mitophagy machinery and eventual engulfment of damaged mitochondria into autophagosomes. PINK1/Parkin-mediated mitophagy ensures the removal of toxic mitochondrial products and prevents neuronal loss. In PD, mutations in PINK1 and Parkin genes result in their loss of function and prevent the damage-sensing mechanism that leads to mitophagy attributed to these proteins.

In healthy mitochondria, PINK1 is constitutively processed by mitochondrial proteases, MPP and PARL, resulting in its proteasomal degradation (left panel). In damaged mitochondria, PINK1 accumulates at OMM bound to the TOM complex where it is activated through dimerization and autophosphorylation. Activated PINK1 subsequently phosphorylates ubiquitin resulting in Parkin activation and relocation to the mitochondria where it further ubiquitinates mitochondrial substrates and signals the mitophagy machinery to remove the damaged organelle.
Abbreviations
PINK1-PTEN-induced kinase 1
MPP-Matrix processing peptidase
PARL-Presenilin-associated rhomboid-like protein
TOM-Translocase of the outer membrane
TIM-Translocase of the inner membrane
OMM-Outer mitochondrial membrane
IMM-Inner mitochondrial membrane
LRRK2
 Since its discovery in 2004, mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have been identified as the most common known genetic contributor to Parkinson's disease. LRRK2 is a large, 265 kD multi-domain protein with both GTPase and kinase function. LRRK2 activity has been associated with a variety of cellular and signaling pathways including mitochondrial function, vesicle trafficking, endocytosis, and autophagy. However, the exact physiological and neurotoxic properties of this protein are poorly understood.
Since its discovery in 2004, mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have been identified as the most common known genetic contributor to Parkinson's disease. LRRK2 is a large, 265 kD multi-domain protein with both GTPase and kinase function. LRRK2 activity has been associated with a variety of cellular and signaling pathways including mitochondrial function, vesicle trafficking, endocytosis, and autophagy. However, the exact physiological and neurotoxic properties of this protein are poorly understood.
LRRK2 PD-associated (LRRK2-PD) mutations are commonly found in its enzymatic core, suggesting that alterations in the function of this region may be responsible for the clinical and pathological phenotypes associated with the disease. The G2019S mutation which occurs in the kinase domain and exhibits increased kinase activity, is the most common pathogenic mutation identified in the LRRK2-PD population. The importance of LRRK2 kinase activity and its connection to PD are highlighted by in vitro overexpression studies where G2019S-LRRK2 was shown to cause cellular toxicity and that inactivation of LRRK2 kinase activity protected cells from neurotoxic insults. Interestingly, LRRK2 was shown to autophosphorylate itself on over 20 serine and threonine residues, adding to the complexity of the potential functional regulation mechanisms of this protein.

Schematic diagram of LRRK2 and its domains. LRRK2 is a multi-domain protein consisting of: 1) an armadillo domain, 2) an ankyrin-repeat region (ANK), 3) an N-terminal leucine-rich repeat domain (LRR), 4) a GTPase domain consisting of Ras of complex protein (Roc) domain, 5) a spacer domain called C-terminal of Roc (COR), 6) a serine/threonine kinase domain, and 7) a C-terminal WD40 repeat domain. The armadillo, ankyrin, LRR and WD40 domains surrounding the core serve as protein-protein interaction domains. The positions of PD-related mutations are indicated with arrows.
LRRK2 & chaperone-mediated autophagy
Impairment of the autophagy pathway, in particular chaperone-mediated autophagy (CMA), is a feature of LRRK2 research models. LRRK2 exerts its toxic effect partly by blocking CMA. Wild type LRRK2 is readily digested by the lysosomes via CMA, however, its disease-causing variants including G2019S-LRRK2 are poorly degraded by this pathway. CMA-mediated degradation of LRRK2 requires binding to cytosolic chaperone hsc70, and recognition of hsc70-LRRK2 complex by LAMP2A on the lysosomal membrane. This interaction leads to the oligomerization of LAMP2A which forms a translocation complex that allows lysosomal import of LRRK2 and its proteolytic degradation within this compartment. PD-related LRRK2 mutants block the oligomerization of LAMP2A and the formation of CMA translocation complex leading to impaired degradation and accumulation of LRRK2 and other CMA substrates including another PD-associated protein, α-synuclein. Interestingly, the toxic effect of LRRK2 on CMA is also manifested in cells that express high levels of the wild type protein, highlighting the need to tightly regulate its intracellular levels.
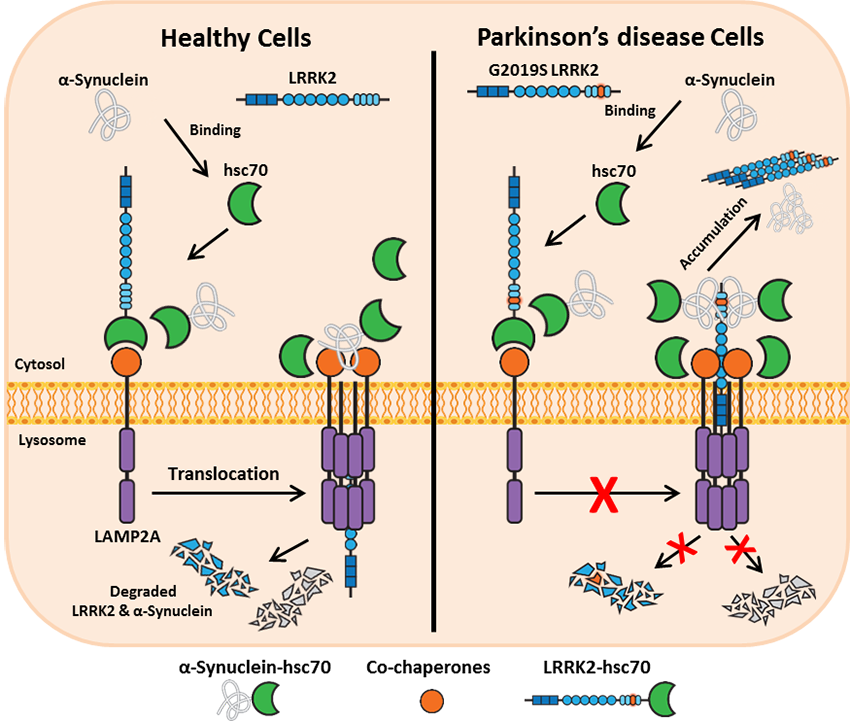
Role of LRRK2 in mitochondrial dysfunction
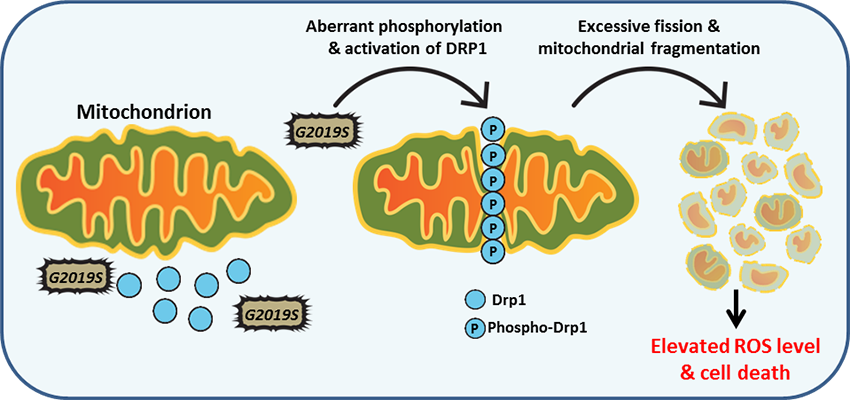
Mitochondrial dynamics are regulated by the function of the mitochondrial fusion and fission machinery. LRRK2 is known to interact with dynamin related protein-1 (Drp1), a key protein involved in mitochondrial fission. Aberrant phosphorylation of Drp1 by G2019S-LRRK2 mutant at the mitochondrial membrane leads to increased fission and build-up of fragmented mitochondria. This in return promotes generation of reactive oxygen species, and ROS-dependent cytotoxicity. These findings suggest that regulation of LRRK2 kinase activity may play an important role in mitochondrial fission events in Parkinson's disease.
BioLegend is proud to offer high quality and specificity reagents for detection of key target proteins involved in the pathogenesis of Parkinson's disease. These reagents include primary antibodies that are highly suitable for use in tissue or cell culture applications, and can be utilized in multiple applications.
LEGENDplex™ Human Neurodegenerative Biomarker Panel 1 (5-plex)
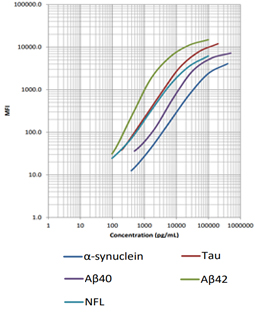
This multiplex bead-based immunoassay utilizes fluorescence-encoded beads suitable for use on various flow cytometers. This panel provides higher detection sensitivity with broader dynamic ranges than the traditional ELISA method. It has been analytically validated and can be used to simultaneously detect five neurodegenerative biomarkers (α-Synuclein, Tau, Aβ40, Aβ42, and NFL) in human biological samples including serum and CSF.
LEGEND MAX™ Human α-Synuclein (Colorimetric) ELISA Kit
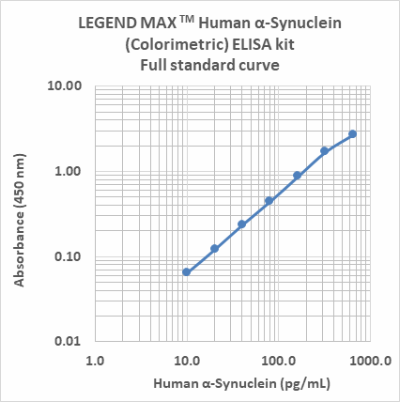
α-Synuclein is an abundant protein in brain neurons where it occurs in multiple forms. The unfolded form is thought to help maintain supply of synaptic vesicles and release of dopamine, a neurotransmitter critical for regulating voluntary and involuntary movements. However, the movement of misfolded α-Synuclein protein between neurons is now believed to underlie pathological PD propagation.
The LEGEND MAX™ Human α-Synuclein (Colorimetric) ELISA Kit is analytically validated with ready-to-use reagents. It comes with a 96-well strip plate that is pre-coated with anti-human α-Synuclein monoclonal antibody. The detection antibody is a biotinylated anti-human α-Synuclein monoclonal antibody. This kit is specifically designed for the accurate quantitation of human α-Synuclein from CSF, serum, plasma, urine, tissue lysate, and cell culture supernatant.
Anti-α-Synuclein Antibodies
We offer purified antibodies specific to α-Synuclein that can be utilized in multiple applications such as Direct ELISA, Western Blot, and Immunohistochemistry. Custom conjugation is available for some products.
Anti-β-Amyloid Antibodies
We offer purified antibodies specific to β-Amyloid that can be utilized in multiple applications such as Direct ELISA, Western Blot, Immunohistochemistry, Immunocytochemistry, and immunoprecipitation. Custom conjugation is available for some products.
Purified anti-α-Synuclein Phospho (Ser129)
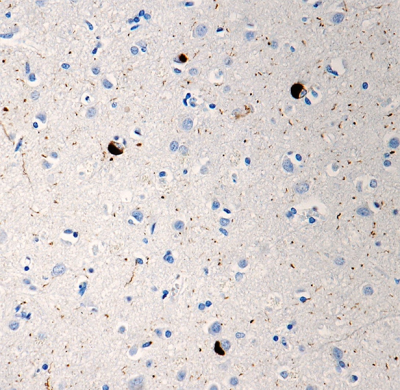
At least three isoforms of α-Synuclein are produced through alternative splicing. The most common isoform is a 140 amino acid-long transcript. Other isoforms include, α -Synuclein-126, lacking residues 41-54; and α-Synuclein-112, which lacks residues 103-130. In addition, α-Synuclein occurs in multiple forms; the aggregate form is one of the major components of inclusion bodies (Lewy bodies) inside nerve cells affected by PD and Lewy body dementia. α-Synuclein is phosphorylated at low levels under normal physiological conditions whereas the majority of the protein is phosphorylated in Lewy bodies at S129.
Purified anti-PINK1

PINK1 (PTEN-induced putative kinase 1) is a 63 kD mitochondrial transmembrane protein that is often cleaved by PARL into a 53 kD fragment. The protein is involved in the clearance of damaged mitochondria via selective autophagy (mitophagy). PINK1 phosphorylation of mitochondrial proteins can protect against mitochondrial dysfunction due to cellular stresses. PINK1 mutations can cause autosomal recessive early-onset Parkinson disease.
Purified anti-DJ-1 (PARK7)

DJ-1 (PARK7) belongs to the peptidase C56 family of proteins and is expressed in almost all human cells and tissues. DJ-1 is a multi-functional protein associated with mitochondrial function, mitophagy, and male fertility. It acts as an oxidative stress sensor, redox-sensitive chaperone and protease. Therefore, the protein has an important role in cell protection against oxidative stress and cell death. DJ-1 is also associated with prostate cancer, and specific mutations of the protein are linked to autosomal recessive, early onset Parkinson’s disease.
ProductsHere



Follow Us