 Login/Register
Login/Register 



Generating the best image involves many factors, all dependent on what is ideal for imaging the marker of interest in its biological context. Here are some considerations when choosing the right reagents and instruments.
Learn more by reading through the topics below or viewing our microscopy FAQs.
Number of Targets
It’s possible to do a five-color imaging experiment with relative ease in both confocal and widefield microscopy. With information about the spectrum of each fluorophore, you can make choices about optimal filter selection to minimize spectral spillover resulting from fluorophores with overlapping excitation and emission spectra. Above five colors, a microscope employing spectral detection becomes useful to unmix the spectral spillover.
Also, if using antibodies for detection, problems can arise with the species-dependence of the primary and secondary antibody combination. Ideally, the use of directly labeled antibodies or haptens like biotin/streptavidin can help.
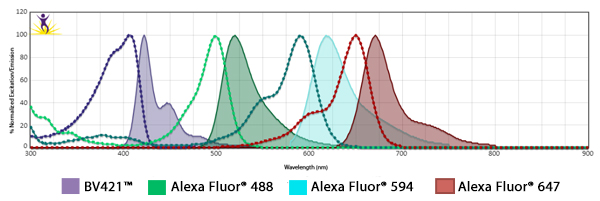
Fluorophore Combinations with Overlapping Spectra
In instances where fluorophores are excited by other wavelengths and have some spillover of their emission into a neighboring filter, the spillover is usually suboptimal strength and results in a weak haze of background. To mitigate this, when using fluors with spillover, ensure the two antibodies are not imaged on markers that co-localize. When possible, one marker could be imaged in the nucleus while the other could be imaged at the cell surface.
In addition, you can make sure the fluorophore that is spilling over into the neighbor filter is on the less abundant antigen. On the subject of filters, they can be optimized to alleviate spillover issues. For example, Alexa Fluor® 647 and Alexa Fluor® 700 share spectral overlap that can sometimes be resolved by narrowing the band pass filter used for detection.
GFP or a Fluorescent Protein Variant
Fluorescent proteins do not survive exposure to methanol or acetone. If the GFP signal was present prior to fixation but the signal is lost upon fixation, check to see if the paraformaldehyde was reconstituted with the help of methanol. If the fixative can’t be changed to be organic solvent-free, anti-GFP antibodies can be employed to recover the GFP signal.
Instrument Choice
The instrument is made to be an ideal tool for the biological question, not the reverse. The better you understand the goal of the image, the better you can match the application to the instrument.
|
Do I want to image tissue thicker than 20 μm? |
→ |
Confocal or Multiphoton Microscopy |
|
Do I want to image more than 4 colors on a cell sample? |
→ |
Spectral Unmixing |
|
Do I want to reconstruct the sample in 3D? |
→ |
Confocal or ApoTome |
|
What level of resolution is desired/ required? |
→ |
Deconvolution, Structured Illumination, STED or PALM |
|
Do I want to demonstrate colocalization/binding or bioactivity in live cells? |
→ |
FRET or FLIM |
Sensitivity
Sensitivity is a balance between the signal strength and non-specific staining/autofluorescence/background. Biological autofluorescence will be endogenous in certain tissues, like brain, liver, lung, etc. The following can be applied to minimize background and improve signal strength:
- An appropriately complex blocking step, for example serum instead of BSA or milk, prior to adding antibodies.
- If streptavidin is used and the tissue will be fixed and permeabilized, an endogenous biotin-blocking kit can prevent the biotin found naturally in mitochondria from binding the streptavidin.
- Use directly conjugated primary antibodies for sufficiently abundant antigens.
- Use secondary antibodies or other amplification methods for lowly expressed antigens. Another option is to use biotin and streptavidin or other hapten-based amplification methods.
- Use enzymatic amplification kits like tyramide signal amplification (TSA) kits for extremely lowly expressed antigens. As a cautionary note, amplification and secondary techniques will also likely increase your background.
Antifade
Mounting media containing antifade is required for the maintenance of signal strength. All organic fluorophores photobleach, a process where reactive oxygen species created in the process of imaging attack the structure of the fluorophores, irreversibly neutralizing their ability to fluoresce. Using antifade is more difficult when the cells are imaged live, since any antifade scavenges oxygen from the media, thus suffocating the cells. This is why regenerating signal, like proteins expressing GFP, are desirable for long-term, live-cell imaging.
Understand “Brightness” in Microscopy
Fluorophore brightness is not a simple measurement of the properties of the individual fluorophore. There are a number of intrinsic and extrinsic factors that affect the final value of brightness. The concepts are explained below.
- The brightness of a fluorophore is the Extinction Coefficient (EC) x Quantum Yield (QY) in water. That’s the output of the fluor. Let’s call it F.
- The antibody has a degree of labeling of the fluorophore; let’s call that DOL. So now brightness is defined as F x DOL.
- You may be able to localize more than one antibody per antigen, as is the case when using secondary detection methods. Let’s call that A. So now brightness is A x F x DOL.
- But ultimately, the brightness of the antibody is determined against the autofluorescence of the background or cells. Let’s call that AUTO. At shorter emission wavelengths (e.g., the FITC channel), autofluorescence can be a massive detractor from the signal. In the near infrared region of a far-red emitting dye, there is hardly any autofluorescence at all. So now brightness can be defined as A x F x DOL - AUTO.
- If you’re comparing two different fluors, it is also important how the pixels in the camera/PMT are binned, i.e., the power of lasers exciting two fluors of different excitation wavelengths and the quantum efficiency of the cameras/PMTs at two very different emission intensities. All these considerations are not universal and extremely variant from instrument to instrument and from end-user to end-user.
The variables listed above do not change the actual brightness of the antibody. Rather, they all affect the brightness perceived by the end user and their ability to image their intended target. The stars in the night sky aren’t dim because of the light pollution of the inner city. The star’s brightness doesn’t change; the conditions to image them do.



Follow Us