 Login/Register
Login/Register 



Multiomics Sample Preparation Guidelines
With the emergence of new single-cell methodologies, it’s important to choose a compatible sample prep strategy. For example, much of our work has focused on the multiomics method known as CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing), which allows for the simultaneous characterization of cell surface proteins with antibody oligo-conjugates and RNA by sequencing. Because you will be relying on antibodies for protein detection in this method, you will want to ensure you use an isolation method that preserves cellular epitopes.
In this blog, we’ll present important technical considerations when planning your single-cell multiomics experiments.
Guidelines for Sample Preparation
The preparation of high-viability single-cell suspensions is absolutely critical to successful single-cell studies because the quality of the sample greatly influences everything downstream and, ultimately, your data quality. You should generate a single-cell suspension that is without clumps and debris and also ensure that you maintain the integrity of the cellular membrane. The exact conditions you’ll need to accomplish this will depend on your sample type and experimental conditions, but, in general, we recommend that you minimize the sample prep time while maintaining cells at a low temperature (on ice). When centrifuging and resuspending the cells, use gentle conditions, such as 300-600 x g centrifuge speeds.
Any isolation and dissociation protocols you use should be optimized to your sample type. For example, if you are isolating PBMCs, you can consider using a density gradient like Lymphopure™ to isolate your cells. Dissociating tissue is more complex, and you’ll likely need to test your protocols to ensure that you’ll still have antibody staining. Try to find a dissociation protocol that uses as gentle lysis conditions as possible (low temperatures and short time periods). To ensure that the cellular epitope hasn’t been altered, you can validate the marker integrity using flow cytometry with a fluorophore-conjugated antibody of the same clone you plan to use in your multiomics experiment.
When possible, try to find a peer-reviewed publication that used an identical or similar sample type to yours in a single-cell experiment to help guide your protocol optimization.
Watch our protocol video to learn how to reliably isolate mononuclear cells from whole blood with Lymphopure.
The Importance of Cell Viability
One of the most important considerations for your sample is cell viability. For a multiomics experiment, you need a sample with high viability (>90-95% is preferred). The presence of non-viable, dying, or clumped cells can lead to:
- An impact on target recovery
- Increased doublets/multiplets ratio
- Release of free-floating, cell-free RNA
- Increased background noise and wasted reads
Remember to check the viability not only immediately after you isolate your samples, but also throughout the experiment. If the viability decreases over a short time period, you’ll see this reflected in your data, often indicated by the presence of high mitochondrial read counts. We also recommend checking your single-cell suspension supernatant for the presence of free-floating RNA, using a product like Ribogreen™, as this RNA can create background noise in all samples and complicate downstream data analysis.
Ultimately, you may not be able to acquire fresh samples with high viability, and you’ll need to remove those dead cells from your sample. If cell death is greater than 30%, we recommend using our MojoSort™ magnetic cell isolation kits for dead cell removal.
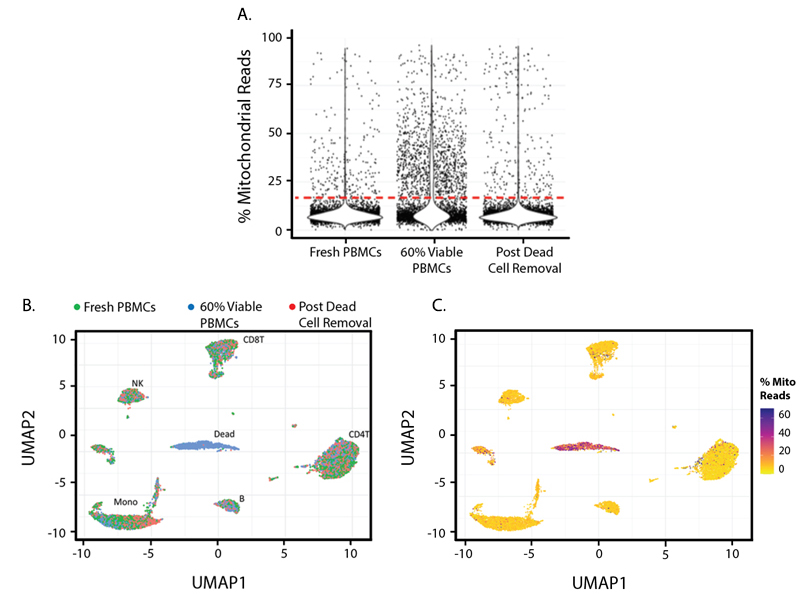
Figure 1. In this experiment, three cell samples were collected and pooled: Fresh PBMCs from a healthy human donor; 60% Viable PBMCs, a cell pool that contains 60% live PBMCs; and Post Dead Cell Removal, which contains the 60% Viable PBMCs pool after they have been cleaned up by the MojoSort Human Dead Cell Removal Kit. Cells with a higher Percent Mitochondrial Read in a library are commonly considered to have low cell viability.
In Figure 1A, you can see mitochondrial reads are more abundant in the 60% Viable PBMCs group and can be removed by the dead cell removal kit. Figures 1B and 1C show UMAPs based on TBNK lineage protein marker expression and demonstrate that common PBMC types can still be detected from all three samples, with the exception of a dead cell cluster in the center of the plot. Figure 1C is colored by percent mitochondrial reads and indicates that the center cluster is comprised of dead and dying cells. This cluster mostly consists of cells from the partial dead cell group with 60% Viable PBMCs.
Another option for improving cell viability in your sample is to use FACS (fluorescence-activated cell sorting). The exact protocol for cell sorting will depend on your cell type and experiment; however, to maintain a high viability sample, we recommend minimizing sorting time, keeping a low flow rate, and using a bigger nozzle, if possible.
Tips for Determining Cell Numbers
Cell number and sequencing depth are dependent on what you’re trying to achieve, your experimental questions, the platform being used, and ultimately, your budget. When helping researchers plan their experiments, the first thing we ask about is their end goals and what tissues/cell types they will be analyzing. For experimental designs with complex heterogeneous cell populations, rare cell types, or cells with low RNA content, larger cell numbers and deeper sequencing will be needed. Focused experiments with homogeneous cell populations can typically use lower cell numbers or less sequencing depth.
If you are simply looking at the differential expression of highly expressed genes between treatments, then larger cell numbers may not be needed and depth can be decreased. In most cases, running small pilot experiments with varying cell numbers and different sequencing depths can help answer these questions. For someone new to the scRNA-seq field, it is important to review the literature and talk with others in the field who have done a lot of the groundwork. There is a balance that needs to be achieved between cell numbers and sequencing depth, and you should not sacrifice one for the other.
Ultimately, preparing a high-quality sample is one of the most important aspects of the single-cell multiomics experiment. While it can seem minor, it requires extensive research and a plan to be in place. A majority of the issues we see in single-cell experiments stem from researchers jumping into experiments without proper planning. Use your resources, consult current literature, talk to other researchers who have done scRNA-seq, and reach out to our technical teams to discuss your experimental questions prior to starting.
While it can seem costly, running small pilot experiments to familiarize yourself with the protocol and identify issues early on will help you avoid wasting reagents, time, and valuable samples.



Follow Us