 Login/Register
Login/Register 



|
|
Please select a category
|

Related Pages
general
FAQs
- Can you provide PDF files for the product related journal articles in general or for the ones you cite on the product TDS?
- We have restricted full journal access. Moreover due to copyright issues we will not send full pdf files for the articles we may have. The end user should do their own independent search to see if the article is provided for free elsewhere.
- How do I know if an antibody is monoclonal or polyclonal?
-
By definition a monoclonal antibody is of one Ig subtype, while a polyclonal antibody contains multiple Ig subtypes. So look at our product isotype description and if it says: IgG1, IgG2a, IgG2b, IgM, etc, these will be monoclonal, whereas: Goat IgG, Rabbit Ig, etc. will be polyclonal. Most of our polyclonal antibodies also have the term poly in the clone name.
- How do I look for reagents that are cross-reactive with a different species (e.g. Rhesus or Cynomolgus)?
-
Perform a search for your species using our search box at the top of the web page. Alternatively, use our advanced search drop down menu for Species Reactivity and select your choice of species. For a broad overview of all our cross-reactive antibodies, use our Antibody Cross-reactivity Chart.
- How do I obtain a Material Safety Data Sheet (MSDS) for your products?
-
On each product data sheet there is a link to all of the available MSDS. Or check our website under "Support". If your product isn't covered in this section, please contact BioLegend Technical Services.
- How long can you guarantee the stability of antibodies when left in ambient temperature?
- The antibodies are fine (unopened in the original box) for up to two weeks at ambient temperature. That being said, we would recommend protecting dyes from extreme temperature and light exposure. If the customer has some doubt about the antibody, it may be best to test it.
- What is BioLegend's quality policy?
-
100% Satisfaction Guarantee. If BioLegend's product does not perform as described on its product data sheet, we'll replace it or refund 100% of the original purchase price
- What is the acceptable tolerance range for storage of antibodies and other reagents recommended at 4°C?
-
There is an acceptable tolerance range between 2°C and 8°C, for all products recommended to be stored at 4°C. If your refrigerator at any time has temperatures outside of this range, we recommend you adjust its settings or use a different refrigerator.
- What is the concentration and expiration date of my antibody lot? Also how can I obtain CoA of the reagent that I received?
-
If you already have purchased the product just simply use the concentration lookup, expiration and CoA related web tools on our website (links below) by typing in the lot# (starts with a letter B) associated with the product. If you have not yet purchased the product then please contact Technical Service for details.
Concentration and Expiration Lookup Tool - What is the epitope of the antibody?
-
BioLegend does not epitope map antibodies. Sometimes this information is indicated on the datasheets, if published, but generally, we will not know the exact epitope of the antibody.
- What is the shelf life of BioLegend products?
-
Most of our products (unless stated otherwise) have a guaranteed shelf life until their expiration date, when under proper storage and handling conditions as instructed on our Product Data Sheets. For recombinant proteins, the minimum guaranteed shelf-life is 6 months from the date of receipt by the end-user.
- Why are some products discontinued?
- Typically the products are discontinued due to low customer demand or their replacement with in-house produced alternatives. In addition, we do side by side comparison of the the new replacement product with the product that is discontinued to make sure it meets our stringent quality control criteria. We don't guarantee discontinued products.
- What is the difference between “Verified” and “Reported” species reactivity noted on antibody product datasheets?
-
Those listed under "verified" reactivity indicate species that have been tested and confirmed to react in-house by BioLegend. Species listed under "Reported" reactivity are those noted by external sources (e.g. literature publications, original antibody developer claims, etc.) but have not been confirmed in-house. Where available, you may be able to find more information by taking a look at the Application Notes and Application References section (under "Product Details") or through literature searches.
ordering
FAQs
- When can I expect to receive my order if I order directly from BioLegend?
-
For U.S. orders, if products are in-stock and your order was made before the shipping cutoff time (contact cs@biolegend.com for more info), they are shipped out that day for you to receive the next day. We typically do not ship on Fridays as to avoid packages sitting unattended over the weekend. International direct orders are shipped on Mondays and Fridays, and typically arrive within 2 - 10 days, depending on how long they are in customs.
- Why should I order from a distributor?
-
For direct orders shipped outside the U.S., shipping costs typically start at USD $75 for antibody orders and $175 for dry ice shipment. This could vary country to country. Additionally, prepayment is required for direct international shipments. Also, taxes and duties will be paid by the recipient upon shipment delivery. In some countries, the customer needs to apply for an import permit. When ordering from a distributor, the payment terms are more flexible, the customer and technical support is more immediate, and the customer has greater convenience in not having to deal with customs agencies, duties/taxes, and currency exchange.
- How are BioLegend reagents shipped?
-
Most products are shipped at ambient temperature. Our products are sufficiently stable to maintain optimal performance after overnight shipping.
- Multiple clones are available for a particular specificity, how do I choose the right one?
-
Check the Product Data Sheets on-line to review applications/characteristics for each clone. If you still have questions, please contact BioLegend Technical Services at:
- Toll Free Phone (U.S., Canada): 1‐877‐273‐3103
Phone: 1-858-768-5801
Fax: 858-455-9587 - E-mail: Click Here
- Toll Free Phone (U.S., Canada): 1‐877‐273‐3103
- What is the advantage of ordering per test instead of per μg?
-
Antibodies offered per test have been pre-titrated for optimal performance in flow cytometry assays. Antibodies offered per plate have been pre-titrated in ELISA. Cell Biology antibodies offered per µl have been pre-titrated for Western Blot. The antibody packed in an μg format (untitrated) is not necessarily always from the same batch number as a test format (pre-titrated) though the same procedures are performed in the quality control testing. Please also note that not all test formats of the antibodies have lower concentrations than the μg format. It depends on a particular batch tested. If the optimal concentration were determined at 1 μg/test for a particular batch, then it would be 100 μg in the 100 tests. The advantage to using a pre-titrated format is that it provides more convenience for the optimal performance in areas such as flow cytometry assays, especially for beginners who may save time and reagents on titration of the antibody.
Flow Cytometry Products
FAQs
- I’ve noticed aberrant signaling in some cell populations of my samples. What could be causing this?
-
Several fluorophores commonly used in flow cytometry and microscopy are coupled to polyethylene glycol (PEG) groups. A significant number of people have detectable levels of anti-PEG antibodies in their blood. At certain concentrations these anti-PEG antibodies can complex to the fluorophore-antibody conjugates leading to aberrant signals in whole blood no wash assays. Beginning in early 2021, we observed an increased frequency of donors showing aberrant staining with PEGylated conjugates in whole blood assays. Preliminary data suggest a correlation between an increased frequency and/or level of anti-PEG antibodies in the donor pool. Some of the COVID vaccines widely administered in early 2021 contain PEG as a component. If your lab conducts experiments employing a whole blood assay, you may encounter such donors. To resolve these cases, we recommend to follow the typical protocol adjustments which are simply adding a wash step, or employing a blocking buffer. For more information please read through our technical note.
- Is the competitor's fix/perm buffer systems compatible with your antibodies for applications such as flow cytometry?
- We have not validated various competitors' fix/perm buffer systems in house so it is empirical to find out if the combination works. It may be best to stick with the buffer system that is validated for a particular clone.
- Is our human Trustain FcX™ (cat# 422302) compatible with anti human CD16, CD32 and CD64 clones 3G8, FUN-2 and 10.1 respectively?
-
Yes
- Is Fc blocking step required for intracellular staining?
- Typically it is not required but if surface staining is involved as part of the staining then do add Fc blocking step.
- Since cyanine-based dyes have a propensity to non-specifically bind to monocyte populations, should I use them for my monocyte phenotyping?
-
This non specific binding should be kept in mind. You can try testing the antibody first to see how much non specific binding you see in your sample. It is also best to follow routine steps such as Fc blocking and titration of the antibody in order to reduce background while doing flow cytometric staining. You can also consider our True-Stain Monocyte Blocker™ to see if it improves staining (Cat. No. 426101).
- How stable is PerCP/Cy5.5 tandem as compared to PerCP alone?
-
PerCP/Cy5.5 is quite photostable and also better than PerCP alone in withstanding fixation.
- How does pH and staining temperature affect Annexin V-Phosphatidylserine binding?
-
Annexin-Phosphatidylserine binding is lost below pH 5.2 and with prolonged incubation over a temperature of 42°C.
- How many biotin molecules are per antibody structure?
- We don't routinely measure the number of biotins with our antibody products but the number of biotin molecules range from 3-6 molecules per antibody.
- I see dim staining with anti human CD357 (GITR) clone 621 in blood and PBMCs.
- Our testing has found that anti human CD357 (GITR) clone 621 stains dim with blood and PBMCs and may only be suitable for situations where GITR levels are high, i.e. stimulated cells or specific cell lines expressing high levels of GITR.
- I see there are multiple clones for the product I want, which one do I choose?
-
If you are not looking for a specific clone check we recommend to use the most popular clone based on Pubmed or Google Scholar. This will give you an idea on how often a clone is used compared to another.
- I am unable to see expression of T cell markers such as CD3 and CD4 post activation.
- TCR-CD3 complexes on the T-lymphocyte surface are rapidly downregulated upon activation with peptide-MHC complex, superantigen or cross-linking with anti-TCR or anti-CD3 antibodies. PMA/Ionomycin treatment has been shown to downregulate surface CD4 expression. Receptor downregulation is a common biological phenomenon and so make sure that your stimulation treatment is not causing it in your sample type.
- I am not getting consistent staining while comparing two different antibody clones from different sources for the same target. What may be the issue?
- Differences in characteristics of antibody clones such as whether they are polyclonal or monoclonal, as well as the source are variables that may lead to inconsistent staining. The difference may be particularly pronounced if a monoclonal antibody is compared with a polyclonal antibody for the same target. Even if the two antibodies are monoclonals, it is possible that their staining pattern may look different. It may be best to search relevant literature.
- I didn't obtain any signal with your fluorescent antibody using compensation beads?
- When using compensation beads please double-check the beads specific details such as exceptions related to certain antibody light chains or host species that may not be compatible with a particular set of commercial beads.
- Which FOXP3 clone should I use?
-
For detecting human FOXP3, both 259D and 206D clones give comparable staining based on in-house testing. Please see the Treg homepage for the differences between clone 206D or 259D. 206D recognizes human FOXP3 epitope in the region of amino acids 105-235. The 259D antibody recognizes human FOXP3 epitope in the region of amino acids 105-235. For other mammalian species, 150D is recommended. Learn more with our Treg webpage.
- What markers would you recommend for my population of interest?
-
Please refer to our Cell Marker webpage . For a more streamlined approach, check out the Essential Markers webpage. It is also advisable to consult the literature to find out further information for a particular cell type.
- What type of PE do you use in your conjugates?
- We use R-PE in our conjugates.
- Will the 0.09% sodium azide in my fluorochrome conjugated antibody affect the viability of my flow sorted cells?
-
Diluting the antibodies to perform the staining would also dilute the azide to a concentration likely not to be toxic to the cells. Since the cells are left in contact with the antibody (and the azide) for only 1/2 hour then washed, the there will be little or no residual azide in the sorting buffer, so the chances of residual toxicity are very low.
- Why does your Anti-Neu5Gc kit (cat# 146901) come with its own blocking solution? Can I use my own?
- Many common blocking solutions contain reagents such as FBS that would contaminate your samples with Neu5Gc, thus leading to generation of background. The blocking solution in our kit is guaranteed to be free of any of these contaminants.
- Why there is not a good correlation between intracellular cytokine staining and ELISA assays?
-
Intracellular cytokine staining for flow cytometry represents a snapshot in time and ELISA represents cytokine accumulation over time.
- Why it is hard for flow cytometry or ELISA compatible antibodies to work for western blot?
- The antibodies that are tested via flow cytometry or ELISA work by recognizing 3D target conformation and epitopes. Harsh treatment associated with WB such as boiling leads to linearization of the target epitopes, which may then lead to non recognition of the target.
- Why is mouse CD206 stained intracellularly and not via surface staining?
-
Typically, mouse CD206 surface level is relatively low under normal conditions and so intracellular staining protocol is required to get better signal.
- What is the concentration of Human TruStain FcX™ (Fc receptor blocking solution, Cat#422301)?
- This information is proprietary.
- What is the molecular weight of your antibodies?
-
We don't test this in house. Typically the molecular weight of an IgG antibody is approximately 150kd.
- What is the most common reason for no IL-17 staining?
-
Most failures of IL-17 intracellular staining are due to stimulation problems. PMA/Ionomycin in liquid form is very unstable. We recommend storing single use aliquots at -20°C degrees. The aliquots should be thawed at ambient temperature and diluted in PBS to a working concentration immediately before stimulation.
- What is the difference between two anti human CD20 clones 2H7 and 1412?
-
For clone 1412: This clone specifically recognizes cytoplasmic domain of CD20 and thus can only be used for intracellular flow cytometry. In this instance you will need to include the fixation and permeabilization steps. Please follow the intracellular flow cytometry staining protocol.
For clone 2H7: This clone is ok for regular surface staining for CD20 and there is no need for any fixation and permeabilization steps.
Our technical protocols can be found here. - What is the difference between test and µg size?
-
The test size products are pre-titrated for optimal staining of 1 million cells in 100 µl volume. On the other hand, µg size products are at a defined concentration regardless of the optimal usage. It is still really important, regardless of format, to use the correct isotype control at the same amount (µg) as their antibody of interest. If the concentration of the antibody is not on the vial, then call us or email (by clicking on “More Info” below) to obtain the concentration for your lot of product.
- What is the F/P ratio for my fluorochrome conjugated antibody?
-
All PE, APC and their tandem dyes labeled antibodies have a F/P of 1:1. Other formats such as FITC or Alexa dyes have various F/P ratios, typically within range of 3-7
- What is the F/P ratio range of our BV421™ format antibody reagents?
-
It is lot-specific. On average it ranges between 2-4.
- Can I use RPMI during Annexin V staining?
-
It is best to follow protocol as described on the product data sheet. Moreover, RPMI 1640 has a relatively high concentration of phosphate and low calcium ion concentration, which negatively impacts Annexin binding to its target phosphatidylserine (PS). Measurement of cell death by using Annexin V may also be significantly affected by time of incubation on ice, calcium concentration, and type of medium.
- Can I use the antibody at a lower concentrate/volume than what is recommended?
-
It is recommend to perform a titration because sometimes you find you can use less antibody for your own experimental needs, and also to have a feel for how the antibody behaves in your own hands in your particular experiment.
- Can I use BioLegend antibodies for cell sorting?
-
In general our antibodies can be used for cell sorting, but we do not routinely test this in house. Please note the fluorochrome conjugates are not endotoxin tested and contain 0.09% azide. Typically this concentration is low, so the azide amount will not affect viability of the cells. As for activation of cells, typically the incubation time with the antibodies for staining is short (~20minutes) and this should not cause activation. Incubation of antibodies with cells at 4°C or on ice will help prevent activation.
- Can I use common compensation control for GFP, CFSE and FITC because they emit in the same channel?
- It is not recommended even if they emit in the same channel because these are still different fluors with different brightness intensities. Individual compensation controls should be employed.
- Can I use less volume or less cells than what is recommended per test?
-
Yes, you can use less cells or less volume per test, as long as the antibody concentration does not change. For example, if you decrease the staining volume from 100 to 50 µl, you could use half the amount of antibody. But, if you decrease your cell numbers from 1 million to 100,000 cells, and your staining volume is still 100 µl, then you should still use the data sheet recommended amount of antibody.
- Do tandem dyes vary between sources and lots?
- No two tandem dyes are created equal. For the same tandem there may be differences between vendors and even between lots. For consistency it may be best to stick with the same source and lot if possible throughout a single study.
- For compensation controls, can I use an antibody with the same tandem dye conjugate but with a different specificity?
-
Since tandems can exhibit variation between sources and lots, it is best to use the exact same antibody as a compensation control. It may be best to use compensation beads for setting up the compensation.
- Can a particular clone work on fixed cells?
-
If the product web sheet for that clone does not reveal this information then it is possible that we have not tested the fixation compatibility of that particular clone. Clone specific literature search is advisable in such situations. Visit our webpage to learn more.
- Can I preincubate my primary and secondary antibodies?
- Theoretically you can if the secondary antibody binds to the Fc portion of the primary. However if the secondary also binds to the light chains then it is possible that it may interfere with the ability of the primary antibody to bind to its target. Therefore it needs to be tested.
- Can I keep my blood sample (drawn in the presence of EDTA) for later analysis by flow cytometry?
- It is advisable to use EDTA blood within 24 hours.
- Can I stain whole blood with anti-FOXP3 using your Foxp3 staining kit?
-
It is not recommended. It is best to use PBMCs for this testing.
- Can FOXP3 be costained with cytokines?
-
The larger holes created by the nuclear permeabilization required for FOXP3 may allow cytokines to leak out of the cell, making it harder to detect lowly-expressed cytokines. You may have to use a control where the cells are only permeabilized through the cell membrane.
- Can I freeze Annexin V conjugates?
-
It should not be frozen as it will lead to loss of biological activity due to dimerization.
- Can I freeze your flow antibodies?
-
We do not recommend freezing our antibodies as this can denature the antibody or cause fluorochromes to uncouple from the antibody during freeze/thaw. In addition, the antibodies can clump and form aggregates, allowing it to bind nonspecifically to cells. We recommend keeping our flow antibodies at 4°C, protected from light.
- Do you sell just the fluorescent dyes separately?
-
We don't offer fluorescent dyes separately. However we do offer custom conjugation services. Please contact our sales department for quotes.
- Does the anti-mouse IL-17 antibody (clone TC11-18H10.1) recognize the A or F isoform?
- Clone TC11-18H10.1 recognizes IL-17A isoform, but it also recognizes the IL-17A/F heterodimer via IL-17A binding.
- Does sodium azide and carrier protein such as serum or BSA interfere with conjugation?
- Sodium azide up to 0.09% w/v does not hinder Amine-Reactive or Sulfhydryl-Reactive conjugations. On the other hand, carrier proteins such as serum or BSA interfere with such conjugations.
- Does staining at room temperature or even at 37°C help for checking chemokine receptors expression?
-
Due to continuous recycling of many chemokine receptors, it may be worthwhile to consider staining at room temperature or at 37°C if the staining at lower temperature (which can potentially reduce receptor turnover) is not optimal.
- Does the signal detected with anti-CD61 conjugates go down as animals age?
-
According to a study (Asselin-Labat, ML et al. 2007. Nat. Cell Biol. 9:201) expression levels of CD61 may vary with age as well as pregnancy stages. It may be best to test CD61 on 4-8 week old mice.
- Do you offer conjugation kits?
- We don't offer dye conjugation kits.
- Do you sell cell lines you use to QC your flow cytometry products?
- We don't offer cell lines. American Type Culture Collection http://www.atcc.org/ may offer further guidance on this issue.
- How does BioLegend choose the optimal concentration of use of an antibody in flow cytometry?
-
Generally, we test 4 to 6 dilutions with the most commonly used target cells (if they are available) for the titration curve, then determine the optimal concentration based on the S/N (Signal/Noise) ratio.
- What is the difference between the FC and ICFC formulation? Are there different optimal concentrations for each?
-
FC is for cell surface staining and ICFC is for intracellular staining. Yes, the concentrations can be different. The IC antibodies and isotypes have optimized fluorochrome conjugation properties to give optimal performance for IC staining.
- How stable is PerCP/Cyanine5.5 tandem as compared to PerCP alone?
-
PerCP/Cyanine5.5 is quite photostable and also better than PerCP alone in withstanding fixation.
- What concentration/amount of the isotype control should I use?
-
Be sure that the isotype control matches the same concentration/amount being used for the primary antibody. Concentrations between isotype and primary antibodies are not always identical. Therefore, using the same volume may not produce equal amounts of antibody.
- I want to stimulate my cells for intracellular cytokine staining. What do you recommend?
-
Please see our stimulation guide.
- I have a custom requests for bulk orders, custom conjugation, custom antibody production, Ultra-LEAF™ formatting.
- Have you tested this Ultra-LEAF™ antibody for in vivo or in vitro applications?
-
We don't test our antibodies for in vivo or in vitro applications unless otherwise indicated. Depending on the product, the TDS may describe literature supporting usage of a particular product for bioassay. It may be best to further consult the literature to find clone specific information.
- How do you determine the antibody concentration?
-
We calculate the concentration by reading the absorbance at 280 nm on a spectrophotometer. The concentration is calculated using the OD280*Dilution Factor/Extinction Coefficient. The extinction coefficient for an antibody with an IgG isotype is 1.4.
- I observed multiple positive peaks when running Compensation Beads. What could be causing this?
-
Multiple positive peaks can occur from insufficient vortexing or mixing of compensation beads. Be sure to vigorously vortex beads for at least one minute before dispensing drops. Longer vortexing helps to disrupt aggregates that can form during storage. Also, ensure that any stray droplets sticking to the side of the tube are thoroughly mixed in with your sample before incubation. If you have already prepared your single-color controls and do not wish to remake them, try setting your positive gate on the major peak, excluding minor stained populations.
Brilliant Violet™
FAQs
- I’ve noticed aberrant signaling in some cell populations of my samples. What could be causing this?
-
Several fluorophores commonly used in flow cytometry and microscopy are coupled to polyethylene glycol (PEG) groups. A significant number of people have detectable levels of anti-PEG antibodies in their blood. At certain concentrations these anti-PEG antibodies can complex to the fluorophore-antibody conjugates leading to aberrant signals in whole blood no wash assays. Beginning in early 2021, we observed an increased frequency of donors showing aberrant staining with PEGylated conjugates in whole blood assays. Preliminary data suggest a correlation between an increased frequency and/or level of anti-PEG antibodies in the donor pool. Some of the COVID vaccines widely administered in early 2021 contain PEG as a component. If your lab conducts experiments employing a whole blood assay, you may encounter such donors. To resolve these cases, we recommend to follow the typical protocol adjustments which are simply adding a wash step, or employing a blocking buffer. For more information please read through our technical note.
- What is Brilliant Violet™?
-
Brilliant Violet™ is a family of highly fluorescent polymers, excitable by the 405 nm violet laser, created by Sirigen based on Nobel Prize winning chemistry. There are seven fluorophores available: BV421™, BV510™, BV570™, BV605™, BV650™, BV711™, and BV785™. Each has a unique emission spectra as well as a unique set of advantages over other 405 nm excitable fluorophores on the market. Brilliant Violet™ fluorophores are suitable for surface or intracellular flow cytometry, providing excellent signal with very little background. For excitation/emission and beta testing data, recommended filters, and comparable flourophores, as well as guidance on the strengths of each member in the family and incorporating them into multicolor panels, see our dedicated Brilliant Violet™ page.
- Can I use all Brilliant Violet™ dyes together?
-
Yes, like for any successful panel, a balance of PMT voltages is beneficial whereby one PMT should not be significantly different from others. Imbalanced PMT voltage situation can potentially amplify the spillover from one fluor to the other. Also pre mixing of the Brilliant Violet™ dyes before staining for an extended time can lead to dye-dye interactions.
- What bandpass filter should I use to detect each Brilliant Violet™ fluorophore?
-
We recommend using the standard 450/50 filter to detect Brilliant Violet 421™, which is the same filter used to detect Pacific Blue™. We recommend using the 510/50 filter to detect Brilliant Violet 510™. For Brilliant Violet 570™ we recommend the 585/42 bandpass filter, commonly used for Pacific Orange™. For BV605™ detection, we recommend the standard Qdot® 605 filter, 610/20 with a 595LP dichroic. For BV650™ detection, the standard Qdot® 650 filter, 660/20 with a 630LP dichroic, is recommended. For BV711™ detection, we suggest the standard Qdot® 700 filter, 710/50 with a 685LP dichroic. For BV785™ detection, we suggest the standard Qdot® 800 filter, 780/60 with a 750LP dichroic. See the Fluorescence Spectra Viewer for complete excitation and emission data.
- How does Brilliant Violet™ perform compared to other fluorochromes?
-
Brilliant Violet 421™ antibody conjugates give an exceptional signal-to-noise ratio, in some cases greater than 10-fold better than Pacific Blue™. It gives PE-equivalent brightness or better on the violet laser. Brilliant Violet 510™ is a non-tandem polymer, and provides improvements over Pacific Orange™, AmCyan, and Horizon™ V500. Brilliant Violet 570™ antibody conjugates also provide much improvement over Pacific Orange™, by as much as 6-fold. Brilliant Violet 605™ and Brilliant Violet 650™ are significantly brighter than eFluor® 605 and eFluor® 650, respectively, by as much as ten-fold, but are not much brighter than Qdot® 605. Brilliant Violet 711™ is significantly brighter than eFluor® 700 and eFluor® 650, by as much as ten-fold, but is not much brighter than Qdot® 705. Brilliant Violet 785™ is similar in brightness to Qdot® 800. Since BV605™, BV650™, BV711™, and BV785™ are non-nanocrystals, they are likely to perform better for intracellular staining and are not affected by fixation. Learn more…
- Can Brilliant Violet™ be used for microscopy?
-
Yes, Brilliant Violet 421™ is exceptionally bright and photostable, making it an excellent choice for microscopy. View the data.
TotalSeq™/Proteogenomics
FAQs
- What are TotalSeq™ reagents and how do they work with established workflows (CITE-seq, and REAP-seq)?
-
TotalSeq™ is BioLegend’s brand of antibody-oligonucleotide conjugates, to enable simultaneous analysis of proteins and mRNA in single cells. CITE-seq and REAP-seq are two similar workflows for simultaneous protein and mRNA analysis, and the TotalSeq™ conjugates integrate seamlessly into these established protocols.
- What is the difference between TotalSeq™-A, -B, and –C?
-
The main difference between the 3 formats relates to the sequences of the oligo tags and their downstream compatibility with single-cell sequencing platforms.
- TotalSeq™-A reagents use a poly-A capture method that is instrument agnostic which includes the 10x Genomics 3’ Single Cell Gene expression kit.
- TotalSeq™-B utilizes the feature barcode technology for 10x Genomics 3’ Single Cell Gene expression kit.
- TotalSeq™-C also uses the feature barcode technology but for 10x Genomics 5’ V(D)J expression kit.
They each use different primers for library amplification. You can read a little more about the differences on our TotalSeq™ webpage. - Can I use TotalSeq™-A and -B together?
-
We are not currently able to support the mixing of TotalSeq™-A and TotalSeq™-B reagents in a single experiment. These two product lines require different protocols for library prep, and the different capturing methods may cause discrepancies in capture efficiency. In addition, it can often cause issues downstream in the bioinformatics portion when analyzing your data due to the variations in index sequences and lengths. In theory, they should work together, but in practice it can be quite difficult and we advise against doing so whenever possible.
- Can I use TotalSeq™-A on the 10x Genomics 3’ Single Cell Gene platform or do I need TotalSeq™-B?
-
Either will work fine.
- What are “features”?
-
When discussing single-cell data, features are any aspect of the cell that is being measured. Common features are mRNA, protein, sgRNA, etc.
- Is TotalSeq™-A, B, and C supported by 10x Genomics?
-
TotalSeq™-A/B/C products are fully supported by BioLegend and have been internally tested by BioLegend on the 10x Genomics platform. TotalSeq™-B and C are fully supported by 10x Genomics. 10x Genomics provides limited support for TotalSeq™-A.
If you are unsure about which technical service team to contact if you have questions or issues, feel free to reach out to BioLegend and we will work with our partners to resolve your problem or answer your technical questions. - What are "Hashtags"?
-
Hashtag reagents are intended to be used for sample barcoding, which allows users to combine multiple samples into a single lane, then de-multiplex during analysis. Instead of select antigen-specific antibodies, the hashtags are designed so that they are specific for human or mouse cells, and to cover as many cells as possible. For human samples, the hashtags are made of two antibodies that recognize ubiquitous surface markers, CD298 and β2 microglobulin, each conjugated to the same oligonucleotide containing the barcode sequence. For mouse samples, the surface markers are CD45 and H-2 MHC class I. The conjugates are already pre-mixed and ready to use.
- What’s the benefit of Cell hashing?
-
Hashing allows customers to run multiple samples in a single lane of a 10x Genomics Chromium instrument, or equivalent, which optimizes the number of cells or samples that can be analyzed simultaneously. This can reduce variability due to batch effects or sample handling. It can also help to identify doublets more efficiently. Furthermore, it can also improve yield and help with cell input when cell numbers are low, therefore optimizing the use of the platform and your workflow or protocol.
- What is “super loading”?
-
Typically, when using a single-cell platform, as you increase the amount of input cells, the rate of doublets (two cells captured as one data point) increases. This leads to an unacceptable percentage of datapoints that contain more than one cell. Hashtags allows you to “super load” the number of cells. To this end, multiple aliquots of the same sample could be stained with as many hashtags as aliquots you’d wish to mix. The number of aliquots and number of cells per aliquot may vary, but after washing and mixing the cells, following the staining and analysis protocol, if more than one hashtag is detected in a single-cell “event”, that event can be safely discarded as a doublet. This does not eliminate doublets that contain the same hashtag but the rate of doublet detection is greatly increased. However, “super loading” should be carefully controlled as the more cells you load the more doublets occur, causing diminishing returns overall. There should be a balance between multi-sample optimization, single-cell data yield, and optimal instrument/platform performance.
- What are nuclear hashtags?
-
Nuclear hashtags are used for single-nucleus RNA-seq (snRNA-seq) samples. snRNA-seq captures RNAs that are isolated with the nucleus. This is done because whole, intact cells may be difficult to isolate due to specific experimental or sample conditions, or to answer a specific scientific question. See an example of nuclear hashtag application in this paper: https://www.nature.com/articles/s41467-019-10756-2
- Can I use nuclear hashtags for single cell ATAC-seq?
-
Unfortunately, our TotalSeq™ reagents are not directly compatible with single ATAC-seq kits at this time. However, the NYGC has pioneered a bridging method ATAC with Select Antigen Profiling by sequencing (ASAP-seq) which is able to bridge TotalSeq™ antibodies with other capture platforms such as scATAC-seq. https://www.biorxiv.org/content/10.1101/2020.09.08.286914v1
Our nuclear hashtag products available off the shelf, but we do not currently have a recommended protocol for this application at this time.
The only recommendations we can provide are literature references such as this one:
https://www.nature.com/articles/s41467-019-10756-2 - Can I get technical support when using hashtags?
-
Hashtags can absolutely be used in any single cell platform including the 10x Genomics platform. BioLegend fully supports the usage of hashtags. Please contact our technical service group for more information.
- Does co-staining for FACS and TotalSeq™ interfere with the workflow?
-
We have not tested this, but there is no reason to believe that non-competing clones for the same target would be problematic. Similarly, a sub-saturating concentration of both reagents against the same target should be tolerated by the cell/technique. The original CITE-seq paper by Stoeckius et al.(Fig. 2) demonstrated that cells can be co-stained for downstream analyses. Although the authors did not use current TotalSeq™ reagents, the technology is the same. In addition, other CITE-seq users have also demonstrated this approach. Please contact our technical service group for more information.
- Why is it essential to remove dead cells prior to subjecting samples for CITE-seq?
-
We recommend >95% viability before beginning you CITE-seq experiment. Running dead cells during CITE-seq essentially leads to “wasting” single cell runs on dead cells, which may ultimately lead to sub-optimal data, or not processing enough cells to pick up the positive events, especially if the frequency of your target cell is very low. Dead cells can also be removed using magnetic beads. If performing CITE-seq before eliminating dead cells is required, it is possible to use bioinformatics methods to try to clean up low quality events (which may include dead cells). This could be a more complex approach, but in such cases, we recommend the following reading:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4758103/
https://www.ncbi.nlm.nih.gov/pubmed/28045081 - Is it necessary to sort samples prior to CITE-seq? What should I consider when deciding to pre-sort, or not?
-
If the populations of interest can be clustered/identified without sorting, the likelihood that you need to pre-sort is minimal. However, if the cell number in the cluster is very small, it may not be sufficient to draw conclusions when compared to control, or other cell populations. In which case, enrichment of this population prior to CITE-seq analysis may help with obtaining more meaningful sequencing data that is selective for your rare cell population(s) of interest. We also recommend to sort out dead cells if the viability in a sample is lower than 95%.
- How can I titrate TotalSeq™ antibodies?
-
We have developed pre-titrated, lyophilized TotalSeq™ panels to facilitate the use of combined antibodies. We have panels in all three formats (TotalSeq™-A, B, and C) designed to identify the main immune cells, as well as a universal panel that contains antibodies against 130 protein targets, and 7 isotype controls. We will keep on releasing these pre-optimized panels as we understand that titrating dozens of antibodies for this application is not an easy task. We may not be able to provide specific titration values for individual antibodies as that may depend on multiple factors. However, here are some recommendations in case you may not be able to use our panels, and need to perform your own titrations:
- Performing titrations using the actual CITE-seq method is costly, but using hashtags can make the process more affordable. See Fig. 3 and associated methods of this publication for more information.
- Perform flow cytometry experiments to find the optimal antibody concentration, using the same clone conjugated to PE. The flow cytometry antibody amount should translate well to CITE-seq.
- Label the cells with different concentrations of a TotalSeq™ antibody, and then use a poly(dT) oligo conjugated to a fluorophore, such as Alexa Fluor® 647, as a secondary reagent to detect the TotalSeq™ antibody. This is a more direct method, but it requires the use of the actual TotalSeq™ antibody.
- What tools are available for data analysis?
-
Some available resources include CITE-Seq-Count, and the tools developed by the Rahul Satija laboratory. Please contact our technical service group for further information.
- I need to order primers; where do I order them and what purity is needed?
-
There are several companies offering primer synthesis. We can recommend IDT technologies. For additive primers, desalt purification is sufficient. For index primers, either HPLC or PAGE is needed.
- Why should I generate ADT/HTO libraries separate from mRNA?
-
Separate ADT/HTO and mRNA libraries provide flexibility when sequencing. Libraries can be mixed at different ratios before sequencing depending on the number of reads needed. Typically, ADT/HTO libraries require less sequencing depth compared to their mRNA counterparts.
- How is the oligo tag bound to the antibody so that it does not interfere with antibody binding to target protein?
-
The oligo is attached to the antibody in the same method as some of our fluorophore-conjugated antibodies that are quality tested by flow cytometry. Once the antibodies have been conjugated to the oligonucleotide, we verify by flow cytometry that the antibody can still bind to its target. This is part of our quality control process for TotalSeq™ conjugates.
- Is it possible to run CyTOF® and TotalSeq™ (CITE-seq) alongside each other?
-
Not in the same experiment; it is not possible because CyTOF® requires its own instrument and protocol, and it destroys the sample in the process. To do so, it is necessary to split the sample and run both CyTOF® and CITE-seq assays. Note that CITE-seq generates equivalent data for surface proteins as CyTOF® with higher panel multiplexing capabilities and at a single-cell level.
- Can CITE-seq be extended to micro-RNAs?
-
This is a technically challenging application as micro-RNAs are very small, and in many cases will be as small as or even smaller than the required primers to execute the protocol. It is not possible at the moment.
- Can I use TotalSeq™ in bulk RNA-seq experiments?
-
Yes, the protocol to perform bulk epitope and nucleic acid sequencing (BEN-seq) has been developed as a collaboration between Illumina and BioLegend. You can read more about it in our application note.
- Is autofluorescence an issue as in flow cytometry?
-
No, sequencing as the final readout is not affected by cell autofluorescence.
- What is the baseline copy number for detection of protein expression (via TotalSeq™ labeling)?
-
This has to do mostly with antibody affinity, titration, and level of antigen expression. BioLegend and some collaborators are working on titration data. There is also some data already published. In theory, as long as the antibody can bind one molecule, PCR amplification and sequencing should pick it up, but there is always background noise. A common way to control for this is to add negative cells to your experiment. For example, if working with human PBMCs, use mouse cells and vice versa.
- How many cells do you need per experiment?
-
This depends on the biological question that needs to be answered. From the technical perspective, when using 10x Genomics Chromium, it is recommended to load 5,000 – 10,000 cells per lane. The use of hashtags can increase that number. For ease of staining, we typically recommend 1 million cells, but this number also depends on availability of cells and the operator’s ability to handle low cell numbers.
- How many TotalSeq™ antibodies is it possible to multiplex?
-
We have optimized multiple pre-mixed antibody cocktails, including TotalSeq™-A, -B, or -C Human TBNK that contain 9 antibodies and our TotalSeq™-C Human Universal Cocktail, v1.0 which contains an unprecedented 130 target antibodies plus 7 isotype controls in a single vial. We will be releasing more panels in the near future. The literature suggests that panels larger than these cocktails are possible. For example, Yapeng et al. published a study using 197 TotalSeq™ antibodies and 7 isotype controls. BioLegend and the scientific community keep pushing the upper limits of multiplexed protein detection by sequencing and have yet to find one.
- Is it possible to do FACS sorting first then do CITE-Seq, while performing both antibody-oligo and antibody-fluorophore labelling at the same time? Will the oligo conjugate withstand sorting?
-
The oligo should withstand sorting. However, based on theoretical considerations we recommend using non-competing clones, to minimize impact in either FACS or sequencing signal/reading. Even if exposed for a very short time, we have not evaluated the effect of ultraviolet or violet lasers on the oligo conjugates. An alternative to flow sorting could be negative magnetic bead enrichment, such as our MojoSort™ platform.
- What should be my sequencing depth be for my ADT/HTO library?
-
We recommend a sequencing depth of 5,000 reads/cell. If you are using more than 100 TotalSeq™ antibodies, you should increase to 10,000 reads/cell. For HTOs alone, 500 reads/cell is sufficient.
- Do I need isotype controls?
-
Isotype controls are highly recommended but not required. The utility of isotype controls in these applications is that they can help identify “sticky cells” caused by cells with compromised viability. Isotype controls also provide an overall picture of the background level or non-specific binding of different cell types. Typically when running isotype controls for CITE-seq assays, you want to include 1 isotype control for every isotype included in the panel, regardless of how many antibodies there are. We recommend using the isotype control at a concentration matching the highest concentration of a single antibody used in a panel, for that particular isotype. Typically, the isotype controls should be used in the same tube as the panel itself. Unlike a traditional flow cyometry experiment, the isotype controls for multiomics cell characterization do not need their own separate samples for comparison. Isotype controls are included in our TotalSeq™ Universal Cocktails.
Another useful control to help identify positive signals are cells known to not bind the antibodies used. For example, murine cells when characterizing human samples, as long as the antibodies do not cross-react between mouse and human targets. This, however, is not required to confidently identify positive signals. - Should I use dextran sulfate to block?
-
Several genomics protocols, including some from 10x Genomics, may recommend including dextran sulfate as a blocker to prevent unwanted charge interactions between nucleic acids and other positively charged molecules. However, we have found that in some cases, the inclusion of dextran sulfate can interfere with antibody binding and recognition and we do not recommend using it with any of our TotalSeq™ reagents. It’s unclear what is the exact mechanism of action that causes this issue. All our protocols reflect this observation; thus we do not recommend the use of dextran sulfate.
- Can I add additional TotalSeq™ antibodies to the TotalSeq™ Universal cocktails?
-
Yes. Additional antibodies can be ‘spiked in’ to your reconstituted cocktail. Please contact our technical service group for recommended guidelines and other considerations.
Covance
FAQs
- Why is BioLegend entering this agreement with Covance?
-
BioLegend is seeking to expand our product portfolio into new areas including Neuroscience, Immunopathology, and IHC reagents. Covance Antibodies not only expand BioLegend's catalog, but also complement current BioLegend offerings.
- Why is Covance selling the Covance Antibody group to BioLegend?
-
The goal of divesting the group was to place it in an organization that better aligns with the focus of the group, would value and grow the group to increase the business. We believe that BioLegend's commitment to providing researchers with the most comprehensive and cutting-edge high quality reagents for life science research, strong background in research tools, and aggressive product development program positions the Antibodies Products group for increased commercial growth and greater career opportunities for employees.
- How does the integration affect my current discounts?
-
Contact our sales team for more detailed information regarding your lab’s discounts.
- If I place an order using the old Covance product name and catalog number, will customer service be able to process my order?
-
Yes, our ordering system will be able to process orders using the old catalog number as well as the new catalog numbers. However, the old catalog numbers will be phased out, so you are encouraged to transition to the new catalog numbers, especially where orders are generated automatically from your systems.
Functional Antibodies
FAQs
- Does BioLegend test each Ultra-LEAF™ antibody by functional assay?
-
No, BioLegend does not test Ultra-LEAF™ antibodies by functional assays unless otherwise indicated. Due to the possible complexities and variations of uses of biofunctional antibodies in different assays and because of the large product portfolio, BioLegend does not currently perform functional assays as a routine QC for the antibodies. However, we do provide references in which the antibodies were used for functional assays and we do perform QC to verify the specificity and quality of the antibody based on our strict specification criteria.
- Does BioLegend test each Ultra-LEAF™ antibody for potential pathogens?
-
No, BioLegend does not test for pathogens in-house unless otherwise indicated. However, we can recommend an outside vendor to perform this testing as needed.
- I am going to do a functional assay. Which grade of purified antibody should I choose?
-
BioLegend's Ultra-LEAF™ (Ultra-Low Endotoxin, Azide-Free) purified antibodies are specifically designed for functional analyses, providing the most accurate results with minimal negative effects. Ultra-LEAF™ antibodies have an endotoxin level < 10 EU/mg, and GoInVivo™ antibodies are pathogen tested and have endotoxin levels < 1 EU/mg of antibody.
- Does BioLegend manufacture antibodies at GMP grade?
-
BioLegend antibodies and reagents that carry the IVD or ASR designation are manufactured in a registered GMP lab, while our other products are manufactured under ISO 13485 certification. Both environments provide high quality products. To learn more about our quality standards visit our Quality Control webpage.
Isotype Controls
FAQs
- IgG2a gene is deleted in some mouse strains, which ones are they?
-
IgG2a gene is deleted in C57Bl/6, C57Bl/10, SJL, and NOD mice. It is replaced with IgG2c gene instead.
- Is it possible to use an isotype control recommended for ICFC for cell surface staining control?
-
It is best to use ICFC validated isotype control for ICFC staining and surface stained validated isotype control for surface staining.
- How do I know how much isotype control to use for test products?
-
Your vial has the antibody concentration on the label, in some cases. If not, contact tech support or use our lookup tool for the concentration of your lot of antibody. Match your isotype to the concentration of the primary antibody.
- How do I choose the right isotype control for the primary antibody I'm using?
-
Refer to the for listed isotype and format of each primary antibody and match the isotype control appropriately. For example, if you have a PE conjugated primary antibody with Rat IgG1, k isotype, use PE Rat IgG1, k isotype control Catalog # 400407 or 400408, as your isotype control. For your conveniece, most products have their isotype control listed under their "related products".
- Can rat IgG1, lambda isotype be substituted with rat IgG1, kappa as an isotype for flow cytometry?
-
Yes, you can since kappa and lambda represent light chains which don't contribute to the background staining.
ELISA
FAQs
- In your LEGEND MAX™ ELISA Kits, there is a step that calls for washing the plates before adding sample. What is the purpose of this step?
-
We typically use a stabilizer for pre-coated plates. The additional washing step is designed to remove these components before you start the assay. If you do not perform the washing, the effect on assay performance is negligible.
- What antibody clones are used in your ELISA Kits?
-
Information regarding the antibody clones used in our kits is proprietary. However, the clonality (polyclonal or monoclonal) and host species of the antibodies may be provided upon request.
- Should I use serum or plasma samples for my ELISA experiment?
-
This is dependent on what targets you are analyzing and the specifics of your study. Ideally, we advise determining the difference between serum and plasma for the targets of interest and deciding on the sample type to be used for quantification. Depending on the targets, there may be a difference in concentrations of the targets between serum and plasma. The most important factor in preparing plasma or serum samples is consistency in preparation to ensure precise measurements. In general, plasma/serum samples should be free of particulate matters, contain no excess lipids, and have no hemolysis.
These types of contaminants will contribute to background, and adversely affect the precision of the assay. The key is to prepare the sample the same way each time. That is, centrifuging samples at the same speed, for the same time, removing the serum or plasma immediately after centrifugation and aliquoting and freezing the samples at the same time. Avoid repeated freezing and thawing cycles. - My sample may have very low levels of analyte. What methods can I use to improve detection?
-
For our LEGEND MAX™ and ELISA MAX™ Deluxe formats we recommend following the provided protocol. We cannot guarantee kit performance if there are deviations from the protocol. Below are general suggestions if you are using our ELISA MAX™ Standard format:
- Control background noise by increasing the number of washings and soaking time between washes.
- Control assay precision. For example, be more careful and more consistent in your pipetting, use fresh paper towels for tapping plates to avoid contamination of avidin-HRP and increase washing volumes.
- Increase incubation times. Be aware that this typically also increases background so the assay sensitivity may not necessarily increase.
- Concentrate your sample, if possible.
- Use a five-parameter logistic curve fitting method to more accurately calculate sample concentration at the lower end of the standard curve.
In many cases, the sample may simply contain very low to non-detectable levels of the analyte. No matter how you manipulate the assay, you may still not be able to obtain detectable concentrations.
- We ran out of capture/detection antibody in our ELISA kit. Can we use a standalone/single antibody to replace it?
-
No, we do not recommend using standalone reagents to replace kit components and cannot guarantee the performance of the kit if you replace or combine reagents from different kits/manufacturers.
- How many samples can I run with your kit?
-
This will depend on whether your samples are analyzed in duplicate or triplicate. For example, after adding the standards, you can run 80 samples with no replicates or 40 samples in duplicate and so on.
- How should the plates be stored after coating?
-
If coated plates cannot be used immediately, they should be sealed and stored overnight at 4°C.
- I have multiple LEGEND MAX™ ELISA kits that I want to run simultaneously. Can I use the same wash buffer for all the kits?
-
The wash buffer provided in all our LEGEND MAX™ kits is the same and the part numbers on the wash buffer bottles in these kits should be identical. For ELISA MAX™ Deluxe and ELISA MAX™ Standard Sets, we provide a recipe for the wash buffer on each kit’s technical data sheet. This recipe is the same for all ELISA MAX™ sets.
- I ran out of assay diluent provided with ELISA MAX™ Deluxe set.
-
You can order more assay diluent using Cat. No. 421203.
- I am using the biotin and purified formats of the same antibody clone to try to set up my own ELISA, but I am having no success.
-
If you are using the same clone for both the detection and capture antibodies, the epitope may already be occupied by one of the antibodies and prevent binding of the other. We recommend choosing different clones for your capture and detection antibodies.
- Where can I find troubleshooting tips for ELISA assays?
-
Please consult our ELISA troubleshooting guide.
- When should I use LEGEND MAX™ ELISA kits with pre-coated plates?
-
LEGEND MAX™ Kits are ready-to-go, designed for ease-of-use, and minimize requirements for coating the ELISA plates and preparing buffer dilutions. We recommend LEGEND MAX™ Kits for users who are new to ELISAs.
- What kind of coating buffer should I use for my ELISA?
-
In general, we recommend using PBS (pH 7.4) or carbonate buffer (pH 9.5). The exact coating buffer you choose may be dependent on the cytokine being detected.
- Why do we recommend not using azide in ELISA buffers?
-
Azide is an irreversible inhibitor of HRP.
- Why there is not a good correlation between intracellular cytokine staining and ELISA assays?
-
Intracellular cytokine staining for flow cytometry represents a snapshot in time and ELISA represents cytokine accumulation over time.
- What is the sensitivity of your ELISA kits?
-
If you are using LEGEND MAX™ or ELISA MAX™ Deluxe format, please consult the individual kit’s manual for more information on sensitivity. We do not provide this information for the ELISA MAX™ Standard Sets but in theory, it should match that of the ELISA MAX™ Deluxe format if you use all of BioLegend’s reagents.
- What is the shelf-life of BioLegend ELISA products?
-
BioLegend's LEGEND MAX™ Kits are guaranteed for 3 months from the date of receipt. ELISA MAX™ Sets are guaranteed for 12 months from the date of receipt. For a lot-specific expiration date, refer to the box label on each product.
- What is the difference between your two substrate solutions: TMB Substrate Reagent Set (Cat. No. 421101) and TMB High Sensitivity Substrate Solution (Cat. No. 421501)?
-
TMB Substrate Reagent Set (Cat. No. 421101) contains two components, which need to be mixed immediately prior to use. The TMB High Sensitivity Substrate Solution (Cat. No. 421501) contains everything in one solution with special stabilizers. It is a high kinetic substrate solution which generally gives higher signal. The type of substrate used for an assay can affect the optical density (OD) achievable and the time required to achieve the desired OD.
- Can I use the recombinant standard provided with the kit for bioassay?
-
No, we do not recommend using the recombinant protein standard for any other applications. The protein standards included in the kit are calibrated for use as an ELISA standard and have been not tested for bioactivity. Additionally, ELISA protein standards may contain other carrier proteins and have not undergone the same sterility testing as our bioactive recombinant proteins.
- Can I use tissue culture grade sterile plates for ELISA?
-
No. Tissue culture plates are designed for cell culture purposes and do not typically have high protein-binding capacity. For ELISAs, we recommend using high protein-binding plates such as Nunc Maxisorp™ plates (Cat. No. 423501).
- Can I use the capture and detection antibody provided as part of an ELISA Kit for other applications such as flow cytometry staining?
-
The antibodies used in our ELISA Kits have been validated for their use in ELISA assays and we cannot guarantee that they will work in other applications, like flow cytometry. Instead, we recommend you use flow cytometry validated antibodies for these experiments.
- Can I use your ELISA kits for my tissue samples?
-
In general, BioLegend’s ELISA products can be used for tissue or cell extracts/homogenates as long as the samples are prepared in such a way that they are compatible with immunoreactivity. For this, we recommend using the following guidelines:
- Tissues/cells should be lysed or homogenized in a neutral pH buffer that contains no denaturing chemicals (such as urea, thiourea, SDS).
- No or minimal levels of detergent (such as SDS, Triton™ X-100).
- No excessive ionic strength (salt concentration greater than physiological ionic strength).
- The buffers should contain sufficient protease inhibitor cocktails to preserve the target proteins from proteolytic degradation by enzymes released from cells.
- Can samples such as serum be reused?
-
We do not recommend reusing samples. For accurate results, samples should be aliquoted and stored for one-time use only. If you decide to reuse a sample, you would need to perform additional validation experiments to ensure you are able to get accurate readings. This would be influenced by a number of factors including sample stability.
- Can I use your matched ELISA antibody pairs with a protein standard from another company?
-
If the antibodies were provided as part of a kit, we do not recommend combining reagents from different ELISA Kits. We cannot guarantee the performance of the kit if you combine reagents from multiple kits or manufacturers. If you have purchased individual antibodies and are developing your own ELISA, it may be possible but would need to be tested. Antibodies may not recognize or show poor binding toward the protein standard if the immunogen used to generate those antibodies is different from the protein standard in terms of sequence or structure.
- At what step can I delay my ELISA procedure for a few days? Can I freeze my plates for later use?
-
It is best to follow the recommended protocol without any additional delays. However, if you wish to pause the experiment, we would recommend delaying the procedure at the blocking step (after coating with the capture antibody). You can add 200 μL of blocking buffer in the wells and keep the plates overnight at 4°C (minimal or no loss of signal) or frozen at -20°C for few days (it may have some impact on the signal). Delaying the procedure at the first step (coating with capture antibody beyond 16-20 hrs. at 4°C) is not advisable because it may lead to high background.
- I ran out of some ELISA kit components, can I them buy separately?
-
It may be possible to purchase some kit components individually. Please contact our technical service group and provide lot information of the kit and its components to determine whether this is feasible.
- Can I mix reagents from different ELISA MAX™ Sets or use them for other applications?
-
This is not recommended. The ELISA MAX™ reagents are optimized for a particular lot, and they have not been quality tested for applications other than ELISA.
- Can coating with capture antibody be carried out for longer than overnight?
-
Generally, coating for 16-20 hours at 4°C is recommended. Longer incubation time may increase the amount of capture antibody bound to the plates and this may increase the background noise.
- Can I add an extra standard at the higher end of the standard curve in order to calculate higher concentration of the analyte?
-
We don’t recommend this practice. Sensitivity of a kit depends on the individual components and their collective validation as a kit and it will not change by adding extra points to the standard curve.
- Can I add an extra standard at the lower end of the standard curve to enhance the sensitivity of my assay?
-
While this may be possible, you may end up with a signal plateau at higher concentrations of the standard. It is generally recommended to use the concentration range recommended in the user manual.
- Do your IL-1β ELISA kits detect pro-IL-1β or the mature form?
-
The antibodies in our IL-1β kits were generated against the mature form of the protein. However, because the pro-form of the protein contains the mature form, the antibodies should detect both forms.
- Does phenol red in the medium interfere with an ELISA assay?
-
No, phenol red in cell culture media will not interfere with the readings from an ELISA assay.
- For some of your ELISA kits, why do my serum samples require dilution with assay buffer?
-
In some cases, dilution with assay buffer is required to minimize the matrix difference between the samples and the standards to achieve better accuracy.
- How can I obtain better signal and sensitivity for my ELISA assay?
-
• Increase incubation times (with samples, detection antibodies, or avidin-HRP or TMB substrate). Please note, this may also increase the background in your assay and may not increase sensitivity.
• Shake plates during incubation steps.
• Ensure that the standard is completely reconstituted before use.
• Increased washing and soaking in between washings to further decrease background.
• If possible, read the plate at 450-570 (or closest) nm for background subtraction.
• Improve duplicate CV% by controlling pipetting error or washing with larger volume of washing buffer.
• Use a 5-PL or 4-PL curve-fitting method, rather than a linear curve fitting. This is usually performed with a curve fitting software and provides better calculation at the lower end of the curve. - What is the difference between your ELISA Kits and Sets?
-
The primary differences between these kits are the reagents included within the kit or set.
LEGEND MAX™: Fully validated and ready-to-use kits. They contain all required reagents, including a 96-well strip plate pre-coated with capture antibody.
RAPID MAX™: cut assay time down to less than 90 minutes. Each kit is fully validated and contains a 96-well strip plate that has been pre-coated to immobilize the capture antibody.
ELISA MAX™ Deluxe: A more cost-effective option that includes most buffers, pre-titrated antibodies, recombinant standard and substrate. Plates are not included in this kit and must be purchased separately.
ELISA MAX™ Standard: Provides the basic components to complete an ELISA including a recombinant protein standard, capture and detection antibodies, and Avidin-HRP. This set requires some optimization and is perfect for those that want to stretch their budget by providing their own buffers and plates.
- Do the ELISA MAX™ Deluxe Sets come with plates?
-
No, ELISA MAX™ Deluxe Sets do not come with plates. Coating Buffer and Assay Diluent (for blocking and dilutions) are included in these sets. Plates can be ordered separately (Cat No. 423501), and instructions for coating the plates are in the sets’ product manuals.
- Can I use different components from different companies for my ELISA?
-
Different manufacturers often use different antibody clones in their kits. The specificity of the antibodies partially dictate how much signal is being detected.
Additionally, different kits may use recombinant protein standards that were expressed and purified using different methods. For example, recombinant proteins expressed in E. coli can show different immunoreactivities primarily due to refolding inconsistences. Kit standards are produced and calibrated against different references between manufacturers, making it difficult to compare components from different kits. Each BioLegend ELISA Kit was developed and validated with reagent concentrations and protocols optimized for analytical robustness. Any changes to the reagents (standards, antibodies, matching matrices) and protocols will affect the final assay performance. - Why are my sample’s cytokine concentrations different using LEGEND MAX™ kits vs other ELISA kits?
-
It is very difficult to obtain exactly the same sample concentrations at pg/ml levels when comparing different cytokine kits from different vendors, in part, because suppliers may use different reagents (including antibody clones) that have different immunoreactivity toward the target protein. It is not uncommon that the cytokine concentration may vary a few-fold between kits.
In general, particularly for those cytokines in pg/mL ranges, it is more important to have matching biological trends instead of matching absolutely concentrations. This is to say, the same cytokine measured by different kits from different vendors should show the same pattern of biological changes for relevant treatments.
- What is the expected concentration of a particular analyte in biological samples (e.g. in serum samples of naÏve animals or normal humans)?
-
Since every sample is unique, it is difficult to predict as this may depend on the sample preparation and the nature of the analyte. If you are using a LEGEND MAX™ Kit, please refer to the respective manual for more information.
- Why can I not connect to the computer or access the software?
-
Make sure that the device is connected to the computer using the micro-USB cable, and that the device has been turned on. Connect the device directly to your computer, do not use an external USB hub. Ensure that the driver for USB connection has been installed.
- Do I need a special power supply?
-
No, the device is connected directly to your computer using the micro-USB cable provided with the device. The device will turn on automatically.
- Is there a power button on the device?
-
No, the device will turn on automatically when it is connected directly to your computer using the provided micro-USB cable.
- Why are signal lights not lighting up?
-
The device has not been turned on. To turn on the device, connect it directly to your computer using the micro-USB cable.
- Why are all signal lights flashing simultaneously?
-
There may be errors related to software or self-test after connection failures. Refer to the troubleshooting section of your manual for error codes and recommended solutions.
- What should we do if calibration fails?
-
Ensure that the slot is empty and that a microtiter plate was not left in the slot during calibration. Ensure that the inside of the slot is not dirty. Refer to the maintenance and cleaning section of your manual for more information regarding how to clean the plate reader.
- What type of microtiter plates can be used for the device?
-
96-well microtiter plates compatible for use with ELISA applications are suitable for use with the Mini ELISA Plate Reader™.
- How can I create a shortcut for the app?
-
Follow the program file path during installation. The shortcut to the app is found in the app folder. You can save a shortcut copy to your desktop.
- For quantitative data analysis, can the fitting method be changed after assay measurement?
-
Yes, it can. Simply open the file containing your saved data, go to set up, and apply the new fitting method. The software will automatically update the results.
- What type of fitting method is used for quantitative data analysis?
-
We recommend using the 4PL regression fitting method with BioLegend ELISA kits. Refer to the ELISA user manual for the recommended fitting method if other ELISA kit suppliers are being used.
- How can I enter the sample dilution factor?
-
Currently, the dilution factor cannot be accounted for while setting up the assay template. The sample dilution factor can be applied manually after the data has been generated.
LEGENDplex™
FAQs
- If I don't have a vacuum, how do I remove the liquid from my plate?
-
If you do not have a vacuum, the assay should be run in a V-bottom plate. After centrifugation using a swinging-bucket rotor with a plate adaptor, you can remove the liquid by flicking the plate quickly, dumping the contents into a sink, and patting it dry carefully on a stack of clean paper towels without losing the beads. Alternatively, you can remove the liquid by using a pipette.
- What are the compatible flow cytometers for the LEGENDplex™ assays?
-
LEGENDplex™ assays can be used on most commonly used flow cytometers that can read APC and PE, such as BD FACSCalibur™, BD FACSCanto™, BD FACSCanto™ II, BD LSR I™, BD LSR II™, BD LSRFortessa™, BD FACSAria™ I, II, III, BD Accuri C6™, BD FACSVerse™, Gallios™, CytoFLEX™, Moflo XDP™, Attune™NxT, NovoCyte™, MACSQuant®, and Guava® easyCyte. Please refer to the “Materials to be provided by end-user” section of your LEGENDplex™ manual for details on the requirements of the machines in terms of laser and channel configurations. For other brands of flow cytometers, the end-user needs to make sure that the machine is set up properly before use.
- Should I perform the assay with the filter plates or with V-bottom plates?
-
Filter plates or V-bottom plates have been included in some kits for your convenience. A vacuum filtration unit is required to work with the filter plates. However, if you don’t have access to a vacuum manifold or if you prefer, then you can use the V-bottom plates and follow the recommended assay protocols for the type of plates you choose. All plates should be made from low binding polypropylene. Polystyrene ELISA or cell culture plates should not be used.
- The kit instruction manual says that the kit is validated for serum, plasma, and culture supernatant sample types. Can I also use tissue homogenates with LEGENDplex™?
-
Yes, all of the above sample types may be used with LEGENDplex™ kits as long as they are prepared in a manner compatible with immunoassays. For tissue homogenate samples, they should be prepared in a neutral pH buffer containing no denaturing chemicals (e.g. SDS, urea, thiourea, cholate, etc.) and no ionic detergents and minimal amounts of non-ionic detergents (e.g. NP-40) may be used in sample preparation. The tissue/cell homogenization buffer should not contain excessive ionic strength above physiological concentrations of salts. If possible, the buffer should also contain protease inhibitors. In addition, the samples should be centrifuged to remove particulates.
- When do I need to do instrument compensation?
-
If a single laser instrument is used for both reporter (PE) and classification (PE/Cyanine5), then compensation is needed to resolve the signal spillover from PE to PE/Cyanine5 and vice-versa. Even on dual laser instruments, some compensation may be required. Please check your machine for the need for compensation. When compensation is required, use the provided calibration beads and follow the Flow Cytometer Setup instructions in the LEGENDplex™ manual. In addition, adjust the PMT voltages such that there is good bead separation, the bead populations are not out of scale, and the PE signal is appropriately amplified to ensure proper signal sensitivity.
- What is the difference between LEGENDplex™ and Luminex® Assays?
-
LEGENDplex™ and Luminex® are both bead-based multiplex immunoassays that use the basic principles of sandwich immunoassays. Both systems use fluorescence-coded beads to achieve multiplexing. The major difference is in the data acquisition. LEGENDplex™ uses common lab flow cytometers for data acquisition, whereas Luminex®-based assays have dedicated machines for this purpose. Therefore, one of the advantages is that LEGENDplex™ can be run on widely available flow cytometers without needing specialized machines. As such, LEGENDplex™ cannot be run on Luminex® machines.
- What curve-fitting algorithm can I use for generating my standard curve?
-
The LEGENDplex™ data analysis software uses a default five-parameter logistic curve-fitting algorithm, which determines sample concentrations and calculates minimum and maximum detection concentrations of the standard curve. If the default algorithm does not work for a particular set of data, other curve fitting methods are available in the software options. If sample concentrations fall outside of the maximum detectable range, the sample will have to be diluted and re-analyzed.
- After I finish the staining process, how long can I wait before reading my LEGENDplex™ samples?
-
The samples can be kept overnight at 4°C while being protected from exposure to light and be read the next day. There may be a decrease in signal, but overall, the assay results should not be affected. Storing the samples for extended periods of time is not recommended, as it could lead to further reductions in signal.
- What is the shelf life of LEGENDplex™ kits?
-
LEGENDplex™ kits are guaranteed for 6 months from the date of receipt, but may have a shelf life of up to 2 years from the date of manufacture.
- Is special software required for data analysis?
-
Typically flow cytometers generate output files in FCS format (e.g. FCS 2.0, 3.0, or 3.1) and in some cases in list mode file format (LMD). Other software may be available to analyze FCS files. Data generated using LEGENDplex™ kits can be analyzed using the freely available LEGENDplex™ data analysis software. Please check our website for the most updated versions of the software.
- My standard curve is not linear; can I use the non-linear part of the standard curve during analysis?
-
Yes. It is possible to use the non-linear part of the standard curve for calculation of results. The LEGENDplex™ data analysis software uses a five-parameter curve-fitting algorithm, which determines the minimum and maximum detection concentrations of each target and reports them. If sample concentrations fall outside of the maximum detectable range, the sample will have to be diluted and re-analyzed.
- Does LEGENDplex™ software run on Macs and PCs?
-
Yes. Our software is compatible with both Macs and PCs. Get more details and download the software at: www.biolegend.com/legendplex/software.
- The biological samples I used in my LEGENDplex™ assay are BSL 2+. Can I fix them prior to flow cytometer acquisition in order to sterilize my samples?
-
The biological samples themselves cannot be fixed before the assay. However, we have found that fixing the beads after the assay with 1-4% paraformaldehyde (PFA) in 1X PBS prior to reading the beads on a flow cytometer is compatible with the assay. Incubation time with PFA should not exceed 10-15 minutes. Prolonged incubation of beads with 1-4% paraformaldehyde will decrease the signal dramatically. Immediately following fixation, the PFA should be washed out using the 1X Wash Buffer supplied with the kit. Beads should be resuspended in the 1X Wash Buffer supplied with the kit.
LEGENDScreen™
FAQs
- If I don't have enough cells and use much less than 4 million/mL (~ 3 x 105 cells/well), will it still work?
-
This may work with lower numbers of total cells, but we recommend trying to keep higher concentrations of cells for faster analysis. Of course, how many cells are needed depends on the specific application. Successful staining has been done with 1 x 105 cells/well.
- What are the guarantees regarding the lyophilized plate compared to the reconstituted plate?
-
Lyophilized product has a guaranteed shelf life of 6 months unopened. Reconstituted plates can be used or stored at 2 - 8°C sealed in the dark and used within a month. Be sure to properly seal the plates to prevent evaporation and shield the antibodies from light.
- How should the kit be stored?
-
The kit should be stored at 2 - 8°C upon receipt. Once opened, the plates must be reconstituted immediately. Reconstituted plates can be used or stored at 2 - 8°C sealed in the dark and used within a month.
- I have added my own antibody solution to the lyophilized product, will the lyophilized antibody work?
-
Yes, as long as the fluorophores on these antibodies are compatible and proper compensation has been applied during acquisition and analysis.
- I am not going to use all the reconstituted antibody solution. Can I keep the left over for later or re-dry the solution in dark?
-
The antibody is in a one test per well format. There will not be any antibody left if the full test is used. Customers may decide to use less than the recommended volume per test, but this is not recommended and the performance is not guaranteed. Customers may also selectively transfer certain antibodies from the original plate to a new plate and use after reconstitution. If any antibody is not used after reconstitution, the plate can be sealed and store at 2 - 8°C for a month in the dark. Once reconstituted, re-drying is not recommended as this may result in a loss of signal.
- Can I use my existing institution discount for this new product?
-
Yes.
- Can I use half or less of the plate and keep the rest for later?
-
Yes. Customers can use half of the plate or whatever specificities they are interested in. However, the whole plate should be reconstituted. The half plate of antibodies must be transferred to another empty plate for the staining. The remaining half must be sealed and stored at 2 - 8°C in the dark and used within a month.
- Are these plates made under sterile conditions?
-
The plates are not sterile. You would handle them as you would handle most typical flow cytometry staining protocols.
- How do I request a custom LEGENDScreen™ product with only my specificities of interest?
-
For more info, visit: biolegend.com/custom_solutions.
- What is the level of variability from one experiment to the other?
-
If the protocol is followed closely, the variability should be minimal. The variability should be similar to single vial antibody staining.
- Is there any data analysis tool or algorithm applicable to this product?
-
Currently, we do not offer any automated analysis solutions for LEGENDScreen™ kits. However, a commercial solution is available from Astrolabe Diagnostics. The Antibody Staining Data Set is an example application of the LEGENDScreen™ Human PE Kit (see Amir et al.).
Live Cell/Dead Cell Discrimination
FAQs
- I am concerned about the spillover I am observing from the Zombie dye into its neighboring channels.
- Rule of thumb with Zombie dyes is to titrate them down as much as possible to fit your application. This should potentially help with spillover. Secondly, Zombie positive events represent dead cells and are typically gated out from analysis.
- How does the performance of your Zombie dye compare with competitors?
-
Zombie dyes have been tested against other leading competitors' fixable viability kits and given comparable results. We also highly recommend that you titrate down the amount of each dye used in order to best match the negative signals of your unstained sample and MFI- (mean fluorescence intensity) stained samples.
- How is your Annexin made and what sequence does it cover?
-
It is made in E. coli, covering human aa Met1-Asp320.
- Why is washing not recommended after the addition 7-AAD or PI addition when assessing viability?
-
These dyes bind in equilibrium with DNA. Therefore, external dye concentration must be maintained during analysis and the dye should not be washed out.
- Can I use methanol/ethanol for fixation after using a Zombie dye?
-
Yes, most fixation reagents are fine to be used with Zombie dyes. However, it should be noted that Zombie dyes can still be sensitive to reactive oxygen species. Light exposure or reagents with hydrogen peroxide can lead to free radical formation, affecting fluorescence.
- Can Zombie be used to determine bacteria, yeast viability?
- We have not tested in house bacterial or yeast viability using Zombie dyes. It is not clear whether the difference between surface and intracellular signals will be significantly different in case of non mammalian cells.
- Can I use Zombie with cells suspension containing serum?
- Serum is full of proteins which will sequester the dye and thereby reducing its effective concentration. The basic rule of thumb with zombie is to titrate it based on your specific condition. Titration also helps reduce the background and spillover into other channels.
- Can I use Zombie dyes for microscopy?
-
Zombie dyes tested in-house for microscopy applications will display data on the product technical datasheet. It should be noted that Zombie dyes may not work for dead cell discrimination in every microscopy application. Important considerations that may impact analysis are determining the signal level that constitutes a dead cell and identifying the proper plane to observe the dead cells.
- Why can't I fix my cells prior to using Zombie dyes?
-
The fixation process can contort and alter the membrane of cells, effectively rendering them dead. Since the ability of the Zombie dyes to stain dead cells is correlated with cell permeability, your results may no longer be a valid representation of dead versus live cells.
- Can I use Zombie dyes to detect apoptotic cells?
-
Yes, Zombie dyes can be used with apoptosis markers, such as Annexin V or Apotracker™ (shown below), to discriminate live, apoptotic, and dead cells.
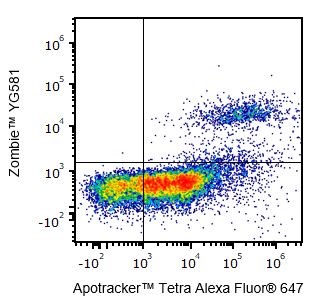
One day-old C57BL/6 mouse thymocytes were stained with Apotracker™ Tetra Alexa Fluor® 647 and Zombie™ YG581. Zombie-dim/Apotracker™-positive cells are apoptotic, while double-positive cells are dead. Live cells are negative for both markers.
- Can I use the UV laser to stimulate Zombie Aqua™? If so, can I then use it in conjunction with BV510™?
-
While we typically do not test Zombie Aqua™ with the UV laser, its excitation peak suggests it is effectively excited at 355 nm. However, we would not recommend using BV510™ off the violet laser and Zombie Aqua™ off the UV laser at the same time. Due to cross-beam excitation of BV510™ by the UV laser and the violet excitation of Zombie Aqua™, this would lead to significantly increased background and excessive compensation requirements.
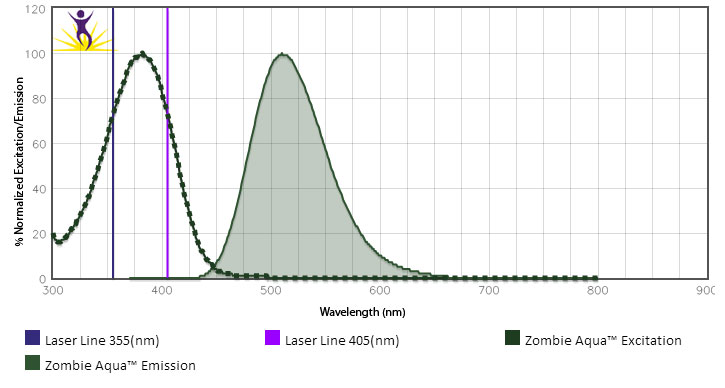
- How should I store Zombie dyes?
-
Store the Zombie dye kit at -20°C upon receipt. Do not open vials until needed. Once DMSO is added, use immediately or store at -20°C in a dry place and protected from light, preferably in a desiccator or in a container with desiccant for no more than one month.
MojoSort™
FAQs
- Is there a way to detach your magnetic particles from the cell surface?
-
No, not currently. We have found that cells are functional without the need to detach the magnetic Nanobeads.
- What is the storage buffer of your particles?
-
The particles are stored in a neutral pH solution containing BSA and sodium azide.
- What is the coating on your magnetic particles?
-
Hydrophilic polymers.
- What is the size of your magnetic particles?
-
The average diameter is approximately 130 nm.
- What is the shelf-life of your magnetic particles?
-
1 to 2 years depending on product type.
- Can your magnetic particles withstand freeze/thaw cycles?
-
Not recommended, however lyophilized particles can be made available as a custom product.
- Are your magnetic particles suitable for use with whole blood?
-
We have cell isolation, selection kits that are pre-optimized for use with whole blood as starting material. In general the MojoSort magnetic particles should be compatible for use in whole blood, but will require optimization.
- Are there any clone restrictions when checking the purity of isolated cells?
-
There are no restrictions for negatively selected cells, as they are untouched by antibodies. For positive selection, there may be clones that are less efficient than others due to possible epitope competition. Please refer to the Technical Data Sheet or contact the Technical Service Group for advice.
- Are downstream applications affected by the magnetic beads bound to the cells?
-
We have tested mouse CD4 positive cells that have been isolated from the spleen and lymph nodes with directly conjugated Nanobeads, as well as CX3CR1 expressing cells from mouse bone marrow. The cells appear unaffected as assessed by migration, differentiation and stimulation assays.
- Can I stain positively sorted cells by flow cytometry?
-
Yes, this is possible, but if you are staining for the same marker as used for sorting, we would recommend using a different clone with non-overlapping epitopes.
- Do your magnetic particles get internalized by the cells?
-
We have not determined this yet.
- Do you provide a custom conjugation service to your magnetic particles?
-
Yes, we do. Please contact our Custom Solutions Team at cst@biolegend.com.
- Do you offer your base magnetic particles (i.e. without any streptavidin or antibody conjugation)?
-
No, not currently.
- Will your magnetic particles separate well in other company's magnetic separation systems?
-
It is possible that other magnetic separation systems can be used. To learn more about this please contact our Technical Service Group.
- What is the composition of your MojoSort™ Buffer?
-
The diluted, 1X MojoSort™ buffer contains 1X phosphate buffer saline (PBS) supplemented with 2 mM EDTA and 0.5% BSA. The 5X solution is sterile. To preserve sterility dilute with sterile water under sterile conditions.
- What antibodies are present in the depletion cocktails provided for isolation kits?
-
Please contact our technical service team for further assistance.
Epitope Tags
FAQs
- What applications can I use epitope tagged antibodies for?
-
The commonly used applications are western blotting, immunoprecipitation and protein purification. These can also be used for immunofluorescence microscopy and flow cytometry.
- Proteins can be detected with anti-protein antibody, but not with the epitope tag antibody. Why?
-
It is likely the target protein was not tagged or the tag is out of the reading frame or the protein is degraded.
- How to set up controls for co-immunoprecipitation experiment?
- For negative control, use an unrelated antibody, or control with the same host species, class, and subclass as the immunoprecipitating antibody. For positive control, use an expression vector with only the epitope tag of interest.
- Where should I put the epitope tag?
- In the literature, the tag has been placed at, or very near, the extreme N or C terminus of the target protein. There are a few reasons behind this choice. Some are historical-like the first proteins to be epitope tagged were tagged at the termini, and when new proteins were tagged it seemed wise to do what had worked in the past. Some of the reasons are practical-tagging is often performed using expression vectors that automatically put the tag at a terminus. And some reasons are theoretical-termini are frequently chosen in the belief that proteins will tolerate additions more readily at these locations than at other sites. While the latter belief may be true-termini are rarely included in active sites, for example-it is also true that many proteins are known for which terminal sequences are critical for function. The termini also appear favorable because they are likely to be on the outside of the folded polypeptide, where one wants the tag to go, and not in the hydrophobic core. But it must be remembered that, due to simple geometry, most of the amino acids in any protein are on the outside, and so, if a protein is tagged at a randomly chosen site, the tag will probably wind up on the outside anyway.
- Will the epitope tag interfere with the function of my protein?
- That has to be determined empirically. However, it is possible that the insertion of the epitope tag may interfere with protein function.
- During Western blot with epitope tag antibodies, there are multiple bands.
-
Several reasons can account for this:
- the early termination of the translation of epitope-tagged protein;
- non-specific detection with anti-epitope tag antibody due to no/poor blocking of the immunoblot.
- partial degradation of the protein
- the concentration of the primary or the secondary antibody is too high.
- Washing between the incubations is not efficient
- How do I introduce the tag?
- The two standard approaches to tagging a cloned gene are: (a) An epitope encoding oligonucleotide is inserted into the coding sequence, or (b) the coding sequence is inserted into an expression vector that already carries the epitope tag. BioLegend does not provide cloning vectors or oligos for cloning. The cloning protocols can be found online or in the literature. When tagging by oligonucleotide insertion, it is important to take into account codon usage preferences for the target cell or organism.
- What are the advantages of Epitope tags?
-
- Tag can be easily and rapidly added to a known gene.
- Multiple tags can be added if required.
- Well-characterized antibodies are available.
- The antibody is specific to the tag, therefore cross-reaction with other proteins is avoided
- Proteins and protein complexes can be purified using standardized practices.
- Tagged proteins can be distinguished from otherwise identical untagged proteins
- Possible to study novel and poorly immunogenic proteins.
- What are the limitations of Epitope tags?
-
A few can be as follows
- A cloned and characterized gene or cDNA must be available
- The epitope tag may interfere with protein structure or function
- Epitope-tagged genes can be expressed at abnormal levels due to the use of heterologous promoters
- The epitope-tagged gene must be introduced into the cell, tissue, or organism of interest.
- Why is there no signal with the epitope tag antibodies on a western blot?
-
A lack of signal following western blotting may indicate a few different problems.
- No or very poor transfer. This can be addressed by quickly checking the membrane with Ponceau-S staining.
- The expression level of the epitope-tagged protein may be too low. Load more ug quantity of the sample and include a positive control.
- A very diluted antibody may be the problem, try a few different concentrations of the antibody to probe the western blot.
- Also, a remote possibility is that the epitope tag is out of frame and is not expressed resulting in a lack of any signal.
NeoClone
FAQs
- Will the prices for former NeoClone products remain the same?
- Yes, BioLegend will not re-price these products. But customers can now use their BioLegend discounts and promotions on former NeoClone products.
- Can I still use the former NeoClone catalog numbers to place my order?
- Customers can find products using the former NeoClone catalog numbers on the website, but all products will be re-assigned a six digit BioLegend catalog number, which will be required for ordering.
- Who do I contact about product questions?
-
Once the products are available on BioLegend's website, contact BioLegend's technical service team for any product questions.
Recombinant Proteins
FAQs
- Why choose BioLegend recombinant proteins?
-
• Each lot of product is quality-tested for bioactivity as indicated on the data sheet.
• Greater than 95% Purity or higher, tested on every lot of product.
• 100% Satisfaction Guarantee for quality performance, stability, and consistency.
• Ready-to-use liquid format saves time and reduces challenges associated with reconstitution.
• Bulk and customization available. Contact us.
• Learn more about our Recombinant Proteins. - How does the activity of your recombinant proteins compare to competitors?
-
We quality control each and every lot of recombinant protein. Not only do we check its bioactivity, but we also compare it against other commercially available recombinant proteins. We make sure each recombinant protein’s activity is at least as good as or better than the competition’s. In order to provide you with the best possible product, we ensure that our testing process is rigorous and thorough. If you’re curious and eager to make the switch to BioLegend recombinants, contact your sales representative today!
- What should I reconstitute the protein with? What do you recommend for its long-term storage?
-
Most of our carrier-free recombinant proteins are shipped in liquid form, so there is no need for reconstitution. If you need to make dilutions, refer to the formulation on the product datasheet. Stock solutions should be prepared at no less than 10 μg/mL in buffer containing carrier protein such as 1% BSA or HSA or 10% FBS (for chemokines, use either BSA or HSA). For long-term storage, aliquot into polypropylene vials and store in a manual defrost freezer. Avoid repeated freeze/thaw cycles.
For reconstitution of our lyophilized recombinant proteins (ELISA Std, animal-free, and some carrier-free) please refer to the Certificate of Analysis and/or Technical Datasheet included with the product and follow the instructions. - What is the difference between carrier-free and animal-free categories of recombinant proteins?
-
Our animal-free products are proteins that go through the entire production process without touching any animal containing components. This includes using animal-free media and purification equipment that are animal component-free. Some studies which are particularly sensitive to contamination by mammalian pathogens may require the use of animal-free products. Our carrier-free products do not contain any carrier protein in the solution, as expected, but they are produced using animal-containing components. Both versions are expected to have similar activity and function, though specific activity is lot-dependent.
- What is a carrier protein?
-
BSA is used as carrier proteins to improve the stability of the reconstituted protein, and to avoid the product from sticking to the wall of the vial.
- What is the specific activity or ED50 of my recombinant protein?
-
The specific activity range of the protein is indicated on the product datasheets. Because the exact activity values on a per unit basis can largely fluctuate depending on a number of factors, including the nature of the assay, cell density, age of cells/passage number, culture media used, and end user technique, the specific activity is best defined as a range and we guarantee the specific activity of all our lots will be within the range indicated on the datasheet. Please note this only applies to recombinants labeled for use in bioassays. ELISA standard recombinant proteins are not recommended for bioassay usage as they are not tested for these applications.
- What is the difference between the carrier-free and the non carrier-free recombinant proteins?
-
All our carrier-free and animal-free formats of recombinant proteins do not have any additional carrier proteins such as BSA in the formulation. Typically our ELISA standard recombinants have carrier proteins added to the formulation for added stability and to avoid the product from sticking to the wall of the vial. When the presence of carrier is not desirable (e.g., in-vivo applications), carrier-free proteins can be used directly. When carrier proteins do not affect the outcome in a study, the customer can decide what type of carrier protein they would like to use and whether it is necessary to add it to their stock.
- Can I use different recombinant proteins from different companies for my ELISA?
-
Antibodies used are different in different kits. The specificity of the antibodies partially dictate how much signal is being detected.
Recombinant standards used are different. First of all, different kits may use recombinant proteins expressed and purified using different method. Second, recombinant proteins expressed from E. coli from the same source can show greater than 10 fold difference in term of immunoreactivity from lot to lot, primarily due to refolding inconsistency. Third, different kit standards can be produced and calibrated against different references. So far there is no universally accepted standardization for cytokine immunoreactivities.
Each BioLegend ELISA product was developed and validated with reagent concentrations and protocols optimized for best analytical robustness. Any changes to the reagents (standards, antibodies, matching matrices) and protocols etc all affect the final assay performance. - Have your recombinants been tested for stability?
-
Our testing shows that the recombinant proteins are able to withstand room temperature for a week without losing activity. In addition the recombinant proteins were also found to withstand four cycles of freeze and thaw without losing activity.
- Do you test the bioactivity of your recombinant proteins with in vivo assays?
- We typically validate the activity of the proteins with in vitro assays as described on the data sheet and not with in vivo testing.
- How are BioLegend's carrier-free recombinant proteins shipped?
-
Our carrier-free recombinant proteins are shipped on blue ice. These products have been validated to maintain activity after shipping using blue ice.
- Does specific activity of a recombinant protein vary between lots?
-
Specific activity will vary for each lot and for the type of experiment that is done to validate it, but all passed lots will have activity within the established ED50 range for the product and we guarantee that our products will have lot-to-lot consistency. Please conduct an experiment-specific validation to find the optimal ED50 for your system.
- How do you convert activity as an ED50 in ng/ml to a specific activity in Units/mg?
-
Use formula Specific activity (Units/mg) = 10^6/ ED50 (ng/mL)
- What is the difference between laboratory (observed) units and international units?
-
There is no direct relationship between International Units and the units that are calculated using the inverse of the specific activity because those units are not calculated using the International Standard provided by WHO (National Institute for Biological Standards and control). In those cases, the best way to compare the activity of two sources of recombinants is by doing the bioassay side by side using the same system. If our protein has been tested using the International Standard, the International Units will be indicated on the technical data sheet for that product.
Western Blot
FAQs
- What are possible causes of High Background in my Western Blot?
-
* Transfer buffers may have become contaminated.
* Post-antibody washes may not have been performed with sufficient time or volume.
* Blocking and incubation agents were not freshly prepared or were too dilute. - What are possible causes of No Signal/Poor Signal in my Western Blot?
-
* Transfer efficiency may have been poor. Check protein transfer by staining the gel and/or membrane.
* Incorrect storage of antibodies or ECL western blotting detection reagents.
* Insufficient protein may have been loaded on the gel. Depending on the location of the target protein, membrane or nuclear preparations may be required (instead of whole cell lysates).
* Film exposure time may have been too short. - How much antibody should I use or what titration range you recommend for WB?
-
0.5 μg/ml to 4 μg/ml range can be used for titrations.
- What is your protocol for treatment of your sample prior to generating the web data?
- Please contact technical service for details.
- You say that the antibody is reported for WB and when I read that reference it didn't provide me with details such as antibody usage amount and other protocol details.
- Unfortunately sometimes the published paper may not have all the necessary details about antibody clone usage. It is best to directly contact the authors for more information. It is recommended to consult additional literature to get further details about the protocol and/or particular cell type or experimental situation. Each experiment is unique and so tissue specific validations and titrations are necessary.
- Why it is hard for flow cytometry or ELISA compatible antibodies to work for western blot?
- The antibodies that are tested via flow cytometry or ELISA work by recognizing 3D target conformation and epitopes. Harsh treatment associated with WB such as boiling leads to linearization of the target epitopes, which may then lead to non recognition of the target.
- What is the molecular weight of your antibodies?
-
We don't test this in house. Typically the molecular weight of an IgG antibody is approximately 150kd.
- If you have not tested/validated the antibody for WB, why do you claim it works for WB?
- We try to provide additional information such as published literature about a particular antibody clone's compatibility with other reported applications so that end users can make an informed decision before embarking on an experiment. It is recommended to consult additional literature to get further details about the protocol and/or particular cell type or experimental situation. Each experiment is unique and so tissue specific validations and titrations are necessary.
- Why is my actual band size different than the predicted molecular weight?
-
The size of the target protein can be affected by many factors such as:
- Post-translational modification such as phosphorylation, glycosylation can increase the size of the protein.
- Post-translation cleavage - e.g. some proteins may get cleaved to the active form from pro-protein form via post translational cleavage.
- Different sized proteins produced from the same gene can be produced by alternative splicing (Splice variants).
- Overall charge on the protein based on amino acid composition can also affect the protein migration in a gel.
- Strong protein-protein interactions can potentially lead to Multimers thereby leading to the appearance of higher size bands. This is usually prevented in reducing conditions and boiling of the sample before loading to the gel.
IHC/IF
FAQs
- You say that the antibody is reported for IHC and when I read your reference it didn't provide me with details like how much antibody to use or detailed protocol.
-
If an antibody is listed as being quality-tested or validated for IHC, we can look up the protocols for you. If this application is reported in literature for IHC, we have not specifically tested the product for this application. It relies on information from publications. Of course, we seek to provide the most updated information on these publications, so we can update our datasheets if the information is incorrect.
- Can I use anti-fade my live cell imaging microscopy?
-
Anti-fade should not be added for live cell imaging as it will deprive the cells of oxygen.
- Can I use GFP expressing cells with Alexa 488 or FITC?
-
No, due to spectral emission overlap you will not be able to distinguish between GFP and AF488/FITC
- Do I need to perform antigen retrieval on my formalin-fixed, paraffin-embedded samples prior to staining?
-
In most cases, this is true. Antigen retrieval helps both the accessibility of the antibody to the tissue and also counteracts the fixation effects on the recognized epitopes. Check the application references for any additional details for IHC or IF experiments.
- Are fluors such as BV570™, BV605™, BV650™, BV711™, BV785™, PE/Cy7, PE/Cy5, APC/Cy7, PE, APC, PerCP, and FITC good for IF?
-
Due to their vulnerability to photobleaching, these fluors are not recommended for IF. Dyes such as Alexa Fluor®, DyLight™, and BV421™ are good alternatives for the IF application.
- Can antibody X be used for immunohistochemistry? What concentration do I use?
-
Typical concentrations of monoclonal antibodies for use in IHC are from 5-25 µg/ml. Polyclonal antibodies can be used at a range of 1-10 µg/ml. We will indicate on the datasheet if an antibody has been tested in-house or published for use in this application. In addition, you can do a lit search with the clone name and immunohistochemistry/paraffin/frozen to see what the protocol details are.
Soluble MHC/Flex-T™
FAQs
- Is there any peptide length recommended?
-
There are no special requirements for the peptides. Peptides that naturally bind to MHC molecules will bind to Flex-T™ reagents. For class I molecules, typical length is about 8 – 10 amino acids. Class II molecules accommodate longer peptides, about 14 – 20 amino acids1.
1) Mohan JF and Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol. 2012 Oct;12(10):721-8. - What are MHC tetramers and what can you do with them?
-
The T-cell mediated innate immune response is defined by the interaction between antigen presenting cells and T cells, through the Major Histocompatibility Complex (MHC) and the T cell receptor (TCR). MHC molecules present a peptide to antigen-specific T cells that recognize this peptide. Soluble, monomeric MHC molecules bind very weakly to the TCR. However, by making a tetramer through a fluorescently labeled streptavidin conjugate, the complex binds to several TCRs, creating a more stable interaction and making it useful for flow cytometric detection of antigen specific T cells.
- I am interested in finding novel peptides instead, are there any resources for this?
-
There are a number of databases and webpages that can help, these are three of them:
- What are the specifications of the UV source?
-
Long-wave UV, 366 nm, 8 Watts (We recommend, for example, CAMAG cat# 022.9115, or Ultraviolet Crosslinker CL-1000). The distance from the solution to the light source should be 2 – 5 cm (approximately 0.8 – 3 inches).
- What is Flex-T™?
-
Flex-T™ (Flexible-Tetramers) is BioLegend’s brand name for our Soluble MHC product line. It encompasses monomers, the ultraviolet (UV) peptide exchange technology, and all associated products and applications.
- Do I need to know the sequence of the UV-labile peptide?
-
The sequence of the UV-labile peptide is not needed to use the reagent. MHC molecules are not stable without a peptide, so these peptides are used just for two purposes: stabilize the MHC molecules and serve as a place holder to be substituted by the peptide of interest.
- Do you offer custom products and services?
-
Yes, please contact our Custom Solution Team with your request at cst@biolegend.com, or contact your local BioLegend representative.
- How do I evaluate the efficiency of the peptide exchange?
-
Follow the protocol for HLA class I ELISA. An assay positive control is provided (Cat#280301) that can be diluted to a high, medium, and low concentration. Signal intensity can be correlated to affinity of the peptide.
- What is the peptide exchange technology and what's the advantage of using it?
-
Flex-T™ MHC monomers are loaded with a peptide that can be degraded by the use of a UV light source. This allows for a peptide exchange when the UV irradiation is done in the presence of the peptide of interest (which is not UV-labile). This flexibility permits the screening of virtually any peptide of interest with enough affinity for the MHC allele that it is loaded onto.
- If I am still interested in "fixed" peptides, what do you recommend?
-
Please see the linked document titled “Peptide Sequences”. The document contains a table with commonly used peptides by allele to study antigen specific T cells.
- Is it feasible to screen not just peptides, but also several specificities of antigen-specific T cells in one sample?
-
With the traditional pre-assembled Tetramer approach, this is difficult to do, and not cost-effective. With Flex-T™ technology, as there is more flexibility to assemble the tetramers, it is easy and affordable to screen a sample for several specificities2. To facilitate this approach, a combinatorial color coding system has been developed. Please visit the Flex-T™ webpage for a detailed description.
2) Hadrup SR et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009 Jul;6(7):520-6. - Is there any advantage in buying biotinylated monomers?
-
Yes, the same monomer can be assembled with different Streptavidin conjugates, providing great flexibility for color choices. Additionally, it can be stored frozen and the tetramer assembled shortly before the experiment. This increases the storage time of the reagent.
Veri-Cells™
FAQs
- Is RNA from Veri-Cells™ able to be used for analysis, for example RT-PCR or RNA flow?
- We have not tried this yet, but it may be possible.
- Is washing required after staining?
-
Yes, it is recommended to wash after staining. We recommend using BioLegend's Cell Staining buffer (Cat. No. 420201) for the wash step.
- How much antibody should I use to stain Veri-Cells™
-
Depending on application pursued it may require diluting the antibodies prior to use.
- What is the expected percentage of CD3+ cells?
- This information is provided in the COA for each lot, it would be similar to what you would see in the “normal” population.
- Can you freeze the cells after reconstitution?
- We do not recommend freezing the cells after reconstitution.
- Can they be used for functional assays?
- This will not work, since the cells are not viable.
- Can the buffer included with the cells be used to keep thawed PBMCs alive?
- We have not tried this.
- Can the cells be activated after rehydration?
- No, they cannot be activated.
- Are these cells virus/pathogen free?
- These are tested to be free of HIV, HBV, syphilis, and HCV.
- Are there any RBCs in Veri-cells™?
- There may be a few, but the majority of RBCs are not present in Veri-Cells™ preparations.
- Are these cells viable?
- No they are not.
- Can a constant percentage of each population be expected in each lot?
- Yes, within each lot, cell populations remain constant.
- Are the cells fixed?
-
They are treated with our proprietary mixture prior to lyophilization.
- Can I treat this like any other sample of PBMCs?
- Yes, make sure to use the Reconstitution buffer for the staining.
- Do Veri-Cells™ stain 100% positive for 7-AAD/DRAQ7?
- Yes.
- Can the cells be used for T or B cell sorting using magnetic bead isolation?
-
Yes, it should be possible, although this has not been verified by BioLegend.
- Can they be used to detect intracellular cytokines such as TNF-α or IFN-γ?
-
For this, use Veri-Cells™ Activated (Cytokine) PBMC which have been activated with Cell Activation Cocktail (with Brefeldin A) containing PMA and Ionomycin. These have been validated to stain positive for activation-induced cytokines including TNF-α and IFN-γ. Veri-Cells™ PBMC and Veri-Cells™ CD4-Low PBMC will not have detectable TNF-α or IFN-γ.
- How do we QC these cells?
-
Each Veri-Cells™ product is tested with a panel of antibodies to ensure expression of these markers falls within the expected range. For more information regarding the exact markers used for each product, please contact technical service.
Enzyme Kits
FAQs
- I have phosphate in my enzyme. What can I do?
-
You can dialyze or desalt the enzyme into a phosphate-free buffer.
- I have 5% DMSO in my assay. Can I use PhosChrome™?
-
Yes, the reagent is designed for drug screening work and other situations that require DMSO.
- Why do I get a high background when my enzyme definitely has no free Pi?
-
This is almost certainly caused by inadequate mixing of the Stabilizer. This results in a high background signal because of non-enzymatic decay of ATP substrate. The Stabilizer is added in a relatively small volume (20μL), and the operation of pipetting up and down with a pipette set to 20μL volume may not result in sufficient mixing when the total volume is 270μL. Try pipetting up and down while stirring at the same time. Alternatively, add the Stabilizer with one pipette set at 20μL volume and mix using a larger pipette set to ~150μL volume. This ensures thorough mixing of the Stabilizer solution with minimal effort.
ChIP
FAQs
- How do I know if my chromatin has been sheared?
-
Purify the DNA from your sheared chromatin samples and run the DNA on a 1-2% agarose gel. DNA should be sheared to a range of 100-900 bp. Below is an example of chromatin from a number of different cells lines that has been sheared optimally using enzymatic digestion compared to chromatin which was over-digested.


Lane 1: Ladder, Lane 2: HeLa, Lane 3: Jurkat, Lane 4: NCCIT, Lane 5: HeLa IL-6 Lane 1: HT1080+ IFNγ, Lane 2: NIH353, Lane 3: HT29, Lane 4: HCT116, Lane 5: Ladder - Which points in the protocol can I stop and save my experiment?
-
Sheared chromatin may be saved at -80°C prior to performing the immunoprecipitation. If you are saving the chromatin, we would recommend aliquoting the chromatin as it can be sensitive to freeze thaw cycles. Alternatively, purified DNA can be saved prior to downstream analysis.
- Should I use sonciation or enzymatic shearing?
-
Sonication may be ideal for difficult to lyse cell types and provides random fragmentation but may damage epitopes. It is not suitable for Native ChIP experiments (in which the chromatin has not been cross-linked). Enzymatic digestion is milder and can be used in Native ChIP experiments but will exhibit sequence bias and may not be suitable for hard to lyse cells.
- What cell types have been validated with the Go-ChIP-Grade™ kit?
-
Human and mouse cell lines such as HeLa, Jurkat, Daudi, THP-1, NIH3T3, and 293T have been validated using this protocol. Other cell types (plant, yeast, tissue, FFPE samples, etc.) may require optimization.
- How do I determine optimal number of cells for ChIP assay?
-
An important consideration when performing ChIP is the amount of chromatin that will need to be loaded to the column in order to elute sufficient IP’ed DNA for downstream analysis. Sufficient cell numbers should be used so that at least 3µg of chromatin can be used per IP. Start with 1-15 million cells. However, the cell number can be scaled up and the reagents need to be adjusted accordingly.
- How much antibody for target protein should be used per ChIP?
-
This should be determined empirically by the end-user. Refer to the datasheet for our Go-ChIP-Grade™ Purified Antibodies, or other ChIP-validated antibodies. The suggested dilution of the Go-ChIP-Grade™ anti-RNA Polymerase II Antibody; Clone 8WG16 (positive control) is 1:300 – 1:500 by volume.
- What is causing high background?
-
The quality of the ChIP antibody has a major impact on the success of the assay. Use only ChIP-validated antibodies. Make sure to have good quality chromatin samples with fragments between 150-900bp. Insufficient DNA shearing leads to longer DNA fragment length that can cause high background in downstream experiments. Insufficient wash steps can also leave traces of non-specific chromatin alongside enriched DNA. If background remains high, include an additional wash step during the IP protocol.
- Why do I not have any enrichment?
-
Make sure to only use ChIP-validated antibodies and follow the instructions for the amount of antibody to be used or titrate the antibody for your experiment. It is also recommended to include ChIP validated positive and negative controls (included in the kit) to validate the efficiency of your ChIP assay. Good quality chromatin samples and chromatin fragment sizes between 150 – 900 bp are also critical. Other factors can include whether cells were effectively lysed, over-fixation and reduced antibody binding due to decreased epitope availability, and insufficient starting material (chromatin sample per IP).
Buffers & Solutions
FAQs
- I’ve noticed aberrant signaling in some cell populations of my samples. What could be causing this?
-
Several fluorophores commonly used in flow cytometry and microscopy are coupled to polyethylene glycol (PEG) groups. A significant number of people have detectable levels of anti-PEG antibodies in their blood. At certain concentrations these anti-PEG antibodies can complex to the fluorophore-antibody conjugates leading to aberrant signals in whole blood no wash assays. Beginning in early 2021, we observed an increased frequency of donors showing aberrant staining with PEGylated conjugates in whole blood assays. Preliminary data suggest a correlation between an increased frequency and/or level of anti-PEG antibodies in the donor pool. Some of the COVID vaccines widely administered in early 2021 contain PEG as a component. If your lab conducts experiments employing a whole blood assay, you may encounter such donors. To resolve these cases, we recommend to follow the typical protocol adjustments which are simply adding a wash step, or employing a blocking buffer. For more information please read through our technical note.
- The Brefeldin A solution freezes at 4°C . Is the solution still OK?
-
We recommend to thaw in warm water after removing from the fridge. The Brefeldin A is dissolved in DMSO, which freezes at 18°C, so this is normal. No one has reported any problems with the freeze/thaw cycles and this is how we recommend it is stored. If customers are concerned, they can aliquot the Brefeldin A to avoid the freeze thaw.
- How long can I store an antibody after I have diluted it?
-
We would not recommend extended storage of the product after dilution. It is best to prepare the dilution as needed on the day of use. Because there are no preservatives in these buffers and they could be susceptible to changes in pH, long term storage greater than a day or two is not recommended.
- When do I use FluoroFix™?
-
FluoroFix™ (cat# 422101) is designed for fixation of cells stained with fluorescent labeled antibodies, including tandem dyes.
- Why do I need to use Annexin V Binding Buffer?
-
Annexin V binding requires the presence of calcium in the solution. So, we provide Annexin V Binding Buffer (cat # 422201), which is optimized for the best performance of Annexin V staining.
- What is the concentration of the 1000X Brefeldin A Solution (420601)?
-
Brefeldin A is provided at a 5mg/ml concentration.
- What is the concentration of the 1000X Monensin Solution (420701)?
-
Monensin is provided at a 2mM solution.
- Can I keep my stained cells in your FluoroFix™ Buffer (422101) for many days before acquisition?
-
Keeping stained cells in any fixative for longer periods of time is not recommended. You can use Fluorofix™ buffer as recommended on the data sheet, followed by washing with cell staining buffer and then at the last step, resuspend your fixed cells in cell staining buffer till you acquire your samples in a day or two.
- Can I stain whole blood with anti-FOXP3 using your Foxp3 staining kit?
-
It is not recommended. It is best to use PBMCs for this testing.
- How do I choose monensin or Brefeldin A solution?
-
Generally, Brefeldin A is more toxic for longer term incubations, so for shorter stimulations (6 hrs or less) use Brefeldin A. For longer stimulations use monensin. We recommend optimization for each cell type and protocol.
Mini ELISA Plate Reader™
FAQs
- Why can I not connect to the computer or access the software?
-
Make sure that the device is connected to the computer using the micro-USB cable, and that the device has been turned on. Connect the device directly to your computer, do not use an external USB hub. Ensure that the driver for USB connection has been installed.
- Do I need a special power supply?
-
No, the device is connected directly to your computer using the micro-USB cable provided with the device. The device will turn on automatically.
- Is there a power button on the device?
-
No, the device will turn on automatically when it is connected directly to your computer using the provided micro-USB cable.
- Why are signal lights not lighting up?
-
The device has not been turned on. To turn on the device, connect it directly to your computer using the micro-USB cable.
- Why are all signal lights flashing simultaneously?
-
There may be errors related to software or self-test after connection failures. Refer to the troubleshooting section of your manual for error codes and recommended solutions.
- What should we do if calibration fails?
-
Ensure that the slot is empty and that a microtiter plate was not left in the slot during calibration. Ensure that the inside of the slot is not dirty. Refer to the maintenance and cleaning section of your manual for more information regarding how to clean the plate reader.
- What type of microtiter plates can be used for the device?
-
96-well microtiter plates compatible for use with ELISA applications are suitable for use with the Mini ELISA Plate Reader™.
- How can I create a shortcut for the app?
-
Follow the program file path during installation. The shortcut to the app is found in the app folder. You can save a shortcut copy to your desktop.
- For quantitative data analysis, can the fitting method be changed after assay measurement?
-
Yes, it can. Simply open the file containing your saved data, go to set up, and apply the new fitting method. The software will automatically update the results.
- What type of fitting method is used for quantitative data analysis?
-
We recommend using the 4PL regression fitting method with BioLegend ELISA kits. Refer to the ELISA user manual for the recommended fitting method if other ELISA kit suppliers are being used.
- How can I enter the sample dilution factor?
-
Currently, the dilution factor cannot be accounted for while setting up the assay template. The sample dilution factor can be applied manually after the data has been generated.
- In what format can the data files be saved?
-
The files can be saved as PDF or CSV. CSV files can be opened by spreadsheet programs, such as Microsoft Excel.
- What types of assays can I run on the Mini ELISA Plate Reader™?
-
We have used the device to perform endpoint ELISA assays with the sample reading at 450 nm and a reference reading at 620 nm. The device contains 4 LEDs: 405 nm, 450 nm, 492 nm, and 620 nm. Any plate-based assay that requires an absorbance reading at one of these wavelengths should be compatible with the plate reader. Additionally, the device provides the option to measure either endpoint and kinetic assays.
- Is there recommended maintenance for this device?
-
The Mini ELISA Plate Reader™ is maintenance-free. There are no parts within the instrument that can be serviced by the user. Each time the instrument is switched on, an internal self-test is carried out to ensure there are no malfunctions.
GMP Recombinant Proteins and Antibodies
FAQs
- What is a GMP recombinant protein or antibody?
-
GMP stands for Good Manufacturing Practice. GMP recombinant proteins and antibodies are manufactured under specific guidelines to ensure product quality and consistency. BioLegend GMP recombinant proteins and antibodies are produced with extensive quality documentation and full material traceability under independent QA oversight.
- What is the difference between BioLegend standard RUO and GMP recombinant proteins and antibodies?
-
BioLegend GMP recombinant proteins and antibodies perform at the same high level as our standard RUO products. No difference in performance should be expected between BioLegend standard RUO and GMP versions. The major difference is the completeness of documentation, material traceability, and independent QA oversight. Additional quality testing may be done depending on the product.
- Are BioLegend GMP recombinant proteins or antibodies animal-free?
-
BioLegend GMP recombinant proteins and antibodies are not animal-free. In all cases where possible, BioLegend attempts to minimize the use of animal components in the manufacturing process. For specific details regarding the use of animal components in the manufacturing process of particular products, please inquire with our technical services team here.
- What is the regulatory status and standards for BioLegend GMP recombinant proteins and antibodies?
-
BioLegend GMP recombinant proteins and antibodies are manufactured in compliance with ISO 13485:2016 and MDSAP. BioLegend GMP recombinant proteins and antibodies are manufactured and tested in accordance with USP Chapter 1043, Ancillary Materials for Cell, Gene, and Tissue Engineered Products and Ph. Eur. Chapter 5.2.12.
Spatial Biology – IBEX
FAQs
- If an antibody clone has been previously successfully used in IBEX in one fluorescent format, will other antibody formats work as well?
-
It’s likely that other fluorophore conjugates to the same antibody clone will also be compatible with IBEX using the same sample fixation procedure. Ultimately a directly conjugated antibody’s utility in fluorescent imaging and IBEX may be specific to the sample and microscope being used in the experiment. Some antibody clone conjugates may perform better than others due to performance differences in non-specific binding, fluorophore brightness, and other biochemical properties unique to that conjugate.
- Will antibodies my lab is already using for fluorescent or chromogenic IHC work in IBEX?
-
Fundamentally, IBEX as a technique that works much in the same way as single antibody panels or single marker IF/IHC. If you’re already successfully using an antibody clone on a sample of interest, it is likely that clone will have utility in IBEX. It is expected some optimization and testing of different antibody fluorophore conjugates will be required to find a suitable format; however, legacy microscopy techniques like chromogenic IHC on fixed or frozen tissue is an excellent place to start looking for useful antibodies.
- Are other fluorophores compatible with IBEX?
-
Over 18 fluorescent formats have been screened for use in IBEX, however, it is likely that other fluorophores are able to be rapidly bleached in IBEX. If a fluorophore format is already suitable for your imaging platform it can be tested for compatibility in IBEX.
- The same antibody works in one tissue type but not another. What is happening?
-
Differences in tissue properties may impact both the ability of an antibody to bind its target specifically and impact the ability of a specific fluorophore conjugate to overcome the background fluorescent signal in a given tissue. Secondary stains, as well as testing multiple fluorescent conjugates of the same clone, may help to troubleshoot challenging targets or tissues. Using a reference control tissue may also give confidence in the specificity of your staining.
- How can I be sure the staining I’m seeing in my tissue is real?
-
In general, best practices for validating an antibody in traditional chromogenic or fluorescent IHC are applicable to IBEX. Please reference the Nature Methods review on antibody based multiplexed imaging for resources on validating antibodies for IBEX.
Flow Cytometry
FAQs
- I’ve noticed aberrant signaling in some cell populations of my samples. What could be causing this?
-
Several fluorophores commonly used in flow cytometry and microscopy are coupled to polyethylene glycol (PEG) groups. A significant number of people have detectable levels of anti-PEG antibodies in their blood. At certain concentrations these anti-PEG antibodies can complex to the fluorophore-antibody conjugates leading to aberrant signals in whole blood no wash assays. Beginning in early 2021, we observed an increased frequency of donors showing aberrant staining with PEGylated conjugates in whole blood assays. Preliminary data suggest a correlation between an increased frequency and/or level of anti-PEG antibodies in the donor pool. Some of the COVID vaccines widely administered in early 2021 contain PEG as a component. If your lab conducts experiments employing a whole blood assay, you may encounter such donors. To resolve these cases, we recommend to follow the typical protocol adjustments which are simply adding a wash step, or employing a blocking buffer. For more information please read through our technical note.
- What is an f:p ratio?
-
Fluorescence to Protein (f:p) ratio tells you the average number of fluorophore molecules that are conjugated to each antibody for any given lot of product. These values can vary widely between manufacturers and even among different lots from the same manufacturer. When using isotype controls, it is important to verify that your control has a similar f:p as that of the primary antibody.
- What controls should I use for a flow cytometry experiment?
-
Isotype controls are recommended for every flow experiment. Isotype controls that do not recognize any known proteins will provide you with information on the background staining of an antibody due to its isotype. For multicolor experiments, fluorescence-minus-one (FMO) controls can help you determine the fluorescence spillover from all your other antibodies and can be used to help in gating positive and negative boundaries. Unstained cells can also provide you with a relative measure of the autofluorescence associated with any particular cell type.
- What is compensation?
-
Compensation is the process of removing spillover (spectral overlap) from other fluorophores into the detector for your fluorophore of choice. For example, due to the wide emission range of FITC, some of the FITC signal “bleeds” into PE giving you false signal in the PE channel. On newer digital instruments, compensation can be applied automatically using single stain controls.
- How does fixation affect fluorophores?
-
Fixation with paraformaldyde generally tends to decrease the fluorescence intensity of bound antibodies, particularly with prolonged exposure. This is especially true for nanocrystals and tandem dyes such as those derived from PE and APC. Excessive fixation can also alter the conformation of proteins, causing loss of antibody reactivity to proteins as well.
- Can cells play a part in specific degradation of tandems?
-
According to a study (Le Roy C, et al. 2009. Cytometry A. 75:882), APC tandems are degraded through a cell-dependent mechanism.
- How are Fluorophores conjugated to antibodies?
-
There are several different methods that can be used to conjugate fluorophores to antibodies. This can depend on the antibody and type of fluorophore being used. One common method is to conjugate the organic fluorophore via primary amines (lysines) on the antibody. Another method conjugates maleimide-labeled fluorophore with antibody via sulfhydryl groups in the hinge region.
- Does this antibody react with extracellular domain or intracellular region of the target?
-
We don't perform epitope mapping in-house. We perform our quality control (QC) test with either surface staining or intracellular staining, as indicated in each product datasheet. When surface staining is performed, it means the antibody recognizes an extracellular epitope. In contrast, if the QC is done with intracellular staining, it means that the protein is expressed in this cellular compartment, but it does not necessarily mean that the epitope recognized is exclusively intracellular. The antibody may still be able to detect the protein if it is expressed or exported to the surface of the cell. It is best to further consult the literature related to the particular clone in this matter.
- How do I know what fluorophores I can use on my instrument?
-
Refer to your instrument manual to determine the specifications of your instrument. You may also be able to find specifications in the software provided with your instrument. Note that many instruments are custom built and may not match the standard manual. Also, many instruments have adjustable filter sets, allowing you to configure your instrument to suitably run many different combinations of fluorophores. Always verify that the instrument you plan to use is capable of detecting the fluorophores in your experimental panel.
- What is fluorescence?
-
Fluorescence is the emission of light by a substance that has absorbed light. In most cases, emitted light has a longer wavelength, and therefore lower energy, than the absorbed wavelength. In flow cytometry, fluorophores are used to tag antibodies in order to provide a signal upon detection of an antigen on the sample when exposed to a single wavelength laser.
- Can I use Maxpar® Ready format clones for flow cytometry staining?
-
We have not tested the Maxpar® Ready antibodies formulated in solution containing EDTA for flow cytometry staining. While it is likely that this will work in majority of the situations, it is best to use the non-EDTA formulated version of the same clone for flow cytometry testing. The presence of EDTA in some situations might negatively affect staining.
- Is there a difference between buffer formulations related to Maxpar® Ready and purified format antibodies?
-
The Maxpar® Ready format antibody clones are formulated in Phosphate-buffered solution, pH 7.2, containing 0.09% sodium azide and EDTA. The regular purified format clones are formulated in solution that does not contain any EDTA. Both formulations are however without any extra carrier proteins.
- How do I know what buffers I need to use for my flow experiment?
-
Be sure to use the buffers recommended in the protocol that you are using. BioLegend provides a number of flow cytometry protocols, as well as a page to guide you in selecting the appropriate buffers for your flow cytometry experiments.
- What is a tandem dye and why do we use them?
-
Tandem dyes are fluorophores composed of two distinct fluorophores conjugated together. The resulting tandem uses the excitation property of the donor fluorophore and emission property of the acceptor fluorophore, based on the principles of fluorescence resonance energy transfer (FRET). This allows for novel fluorophores with high stokes shifts (separation between excitation and emission range).
- Can I swap filters from one laser line to another in a FACS machine?
-
If you swap the filter then it may be best that you test the settings with multipeak (6-8) beads such as our Rainbow Calibration Particles and look at their spread with the original filter and recheck the bead spread with the changed filter.
- Do you guarantee that your antibodies are totally pathogen free?
-
BioLegend does not test for pathogens in-house aside from the GoInVivo™ product line. However, upon request, this can be tested on a custom basis with an outside, independent laboratory.
- I observed multiple positive peaks when running Compensation Beads. What could be causing this?
-
Multiple positive peaks can occur from insufficient vortexing or mixing of compensation beads. Be sure to vigorously vortex beads for at least one minute before dispensing drops. Longer vortexing helps to disrupt aggregates that can form during storage. Also, ensure that any stray droplets sticking to the side of the tube are thoroughly mixed in with your sample before incubation. If you have already prepared your single-color controls and do not wish to remake them, try setting your positive gate on the major peak, excluding minor stained populations.
Flexi-Fluors
FAQs
- What are Flexi-Fluors?
-
Flexi-Fluors are rapidly made-to-order conjugated antibodies. The technology, manufacturing processes, and specifications used to create Flexi-Fluors are the same as our regular catalog products. However, the optimal concentration and performance of each Flexi-Fluor must be determined by the customer.
- How quickly will I receive my order?
-
We aim to ship Flexi-Fluors within 2-3 weeks of receipt of your order. However, depending on your location, shipping times may vary.
- How are Flexi-Fluors different from regular catalog products?
-
Flexi-Fluors are made on demand, specifically for you. Flexi-Fluors are manufactured using the same high-quality standards, and specifications as other catalog products. For faster delivery, Flexi-Fluors are not tested by flow cytometry to determine optimal concentrations or evaluate performance. This testing needs to be performed by the customer.
- How do I determine the optimal concentration for using my Flexi-Fluor? How should I titrate my antibody?
-
Flexi-Fluors are provided at a standard 0.2 mg/mL concentration. We recommend that you titrate your antibody to determine the optimal concentration to use for your application. For many flow cytometry applications, conjugated antibodies perform well at concentrations ranging from 0.03 to 1.0 µg per million cells in 100 µL volume. We recommend that you test a range of concentrations starting from 10 µg/mL.
For example, make five 1:1 serial dilutions of your 0.2 mg/mL antibody. Add 5 µL of each dilution (including the undiluted antibody) to 100 µL of cells (at 107 cells/ml) to test six concentrations - 1.0, 0.5, 0.25, 0.125, 0.06, and 0.03 µg per million cells in 100 µL volume. Compare staining patterns or create a titration curve using the MFI or staining index to determine the optimal concentration.
- I can’t find the antibody-dye combination that I need. When will it be available?
-
We continuously update our catalog, introducing scores of new products every month. Please get in touch with our Technical Service team for an update on new products or recommendations for suitable alternatives to complete your panel. Or contact Custom Solutions to inquire about our affordable custom conjugation services.
- I need help to validate the performance of my Flexi-Fluor. Who should I contact?
-
Please get in touch with Technical Service for assistance.
- Can I order more than 50 μg of a Flexi-Fluor?
-
Yes, you can order multiple vials of the same Flexi-Fluor products. We cannot guarantee, however, that these vials will be bottled from the same lot. For bulk single-lot orders, contact our Custom Solutions team.
- What is the expiration date of my Flexi-Fluor?
-
Expiration dates can be found on the vial label or by using our CoA lookup tool.
ProductsHere



Follow Us