 Login / Register
Login / Register 



Microglia are a glial cell type residing in the central nervous system (CNS) and are recognized for their role in CNS development (e.g.synapse remodeling and maturation), maintenance of homeostasis, and involvement in degenerative diseases. This page provides background detail about microglia and mechanisms by which they contribute to disease. BioLegend offers a wide range of products including antibodies, immunoassay solutions, and magnetic cell separation kits to identify and study microglia.
Loading...
Microglia
Microglia are the resident macrophages in the CNS, and are ontogenically distinct from other phagocytic cells in the mononuclear phagocyte system such as dendritic cells, macrophages and monocytes. Microglia originate from primitive macrophages derived from erythro-myeloid precursors in the yolk sac. These cells colonize the brain during development and remain of embryonic origin in the healthy CNS. In the steady-state condition, microglia can be identified using several common markers which they share with macrophages, such as CD11b, CD45, CD68 and CX3CR1. These cells also express specific markers such as P2RY12, which allow for their distinction from other cell types in the brain as well as peripheral immune cells.
Microglia actively survey their surrounding domain and can rapidly respond to environmental changes such as an immune threat. As a consequence, these cells undergo morphological changes to take on an amoeboid shape and become phagocytic to remove the encountered threat. Under inflammatory conditions, microglia upregulate markers such as CD45 and MHC class II. These morphological changes are also accompanied by expression and secretion of inflammatory molecules such as cytokines and chemokines, which help microglia communicate with astrocytes and peripheral immune cells.
Synapse loss is a common feature of Alzheimer's disease (AD) and often correlates with cognitive dysfunction and decline. Deregulation of the complement cascade, which plays an essential role in innate and adaptive immune responses, has been suggested as a contributing factor leading to chronic inflammation and neurodegeneration observed in AD. Supporting evidence demonstrate localization of a full range of complement proteins from C1q to C5b-9 (membrane attack complex) with amyloid deposits as a prominent feature of AD lesions (1). Notably, mRNA levels of complement components are upregulated in the affected brain regions of AD patients (2). Research has shown that amyloid-beta (Aβ) aggregates can initiate antibody-independent complement activation by binding to C1q and C3b (1). Furthermore, Aβ deposits are frequently surrounded by reactive microglia and astrocytes which are capable of producing complement proteins as well as phagocytosing and degrading Aβ (3). These inflammatory responses appear to initially play a protective role by removing Aβ deposits. However, the continuous generation of Aβ aggregates leads to persistent complement activation and sustained inflammation that is detrimental to neuron health in the brain.
Recent findings in AD-like mouse models suggest that in early stages of AD, corresponding to the pre-plaque phase of the disease, C1q can tag synapses for engulfment by microglia. Aberrant synaptic pruning as a result of an interaction between Aβ, C1q and microglia may play an important role in excess synapse loss and dysfunction prior to plaque formation in AD (4).

Schematic representation of Aβ-mediated synapse loss. In early stages of AD, Aβ oligomers lead to synapse engulfment and removal through interactions between C3, and its receptor CR3 expressed on microglia. At the late stages of disease, localization of microglia with amyloid plaques become apparent.
References:
Neuroinflammation is a prominent feature of Parkinson's disease (PD) and can contribute to disease progression through loss of dopaminergic (DA) neurons in the substantia nigra (SN) of the brain. Elevated levels of pro-inflammatory cytokines and extensive proliferation of reactive microglia around dopaminergic neurons have been detected in the SN of post-mortem brains from PD patients (1). Notably, microglia are enriched in SN compared to other regions of the brain. These findings, coupled to reduced antioxidant capacity and enhanced sensitivity of neurons to pro-inflammatory molecules, support a role for microglia-mediated DA degeneration in PD.
The leucine-rich repeat kinase 2 (LRRK2) protein may act as a mediator of inflammation through cytokine signaling and microglia-mediated pro-inflammatory responses. Mutations in the LRRK2 gene are the most common cause of familial PD. LRRK2 is a multi-functional protein with ubiquitous expression; its mRNA and protein levels are highly expressed in immune and glial cells, and neurons (1). Recent findings indicate the presence of LRRK2 splice variants in the brain with neurons and astrocytes expressing a different variant than microglia, suggesting a differential function for this protein in various cell types (2). In line with these observations, lipopolysaccharide (LPS)-induced inflammation in mice was associated with upregulation of LRRK2 protein expression and activity in microglia (3). Furthermore, down-regulation of microglial LRRK2 protein expression attenuated LPS-induced inflammatory responses through pro-inflammatory cytokine signaling. Interestingly, gene expression profiling of microglia from mice that over-express LRRK2 R1441G, a disease-associated mutation, revealed an expression profile corresponding to reactive and pro-inflammatory phenotype under normal, unstimulated conditions (1). These combined findings indicate a role for LRRK2 in cellular pathways that induce inflammation and that its dysfunction may exacerbate microglial pro-inflammatory responses that would ultimately lead to neurodegeneration.
References:
1. Russo I, et al. 2014. J. Neuroinflammation. 11:52. Pubmed
2. Giesert F, et al. 2013. PLoS One. 8(5): e63778. Pubmed
3. Moehle MS, et al. 2012. J. Neurosci. 32(5):1602. Pubmed

Abnormal LRRK2 activity affects microglia function by inducing pro-inflammatory phenotype in these cells, exacerbating the inflammatory responses, and ultimately leading to neuronal loss observed in PD.
Under injury-associated conditions, ATP released from the damaged tissue or blood vessels elicits an astrocyte-derived ATP gradient, which is sensed by microglial P2RY12. This gradient triggers microglia to elongate their processes and move towards the site of injury. Rapid microglial process extension also helps cover the injured capillaries and seal the breached area.
Injury-activated microglia can also secrete IL-1α, tumor necrosis factor, and C1q to induce an A1 phenotype in astrocytes where they lose their normal synaptogenic and phagocytic functions, gaining cytotoxic ability to kill neurons and oligodendrocytes. In this context, the microglia-astrocyte interaction may help drive neurodegeneration.


(Left) In response to blood vessel injury, astrocytes generate an ATP gradient, which leads to microglial process elongation and chemotaxis towards the site of injury to help seal the injured site. (Right) Damage-associated ATP can also trigger microglial-induced reactive astrocytes with cytotoxic phenotype and ability to kill other neuronal and glial cell types.
References:
1. Li Q. and Barres B. Nat. Rev. Immunol. 10.1038/nri.2017.125. Pubmed
BioLegend offers a wide range of high quality and specificity products to study microglial cells. These reagents include antibodies, immunoassay solutions, and magnetic cell separation kits that enable research into microglial morphology and function utilizing multiple applications.

Purified anti-CX3CR1 Antibody (clone 8E10.D9)
Clone 8E10.D9 demonstrates specificity for human CX3CR1 and is effective for use in IHC and WB. This clone is available in biotin-conjugated format.
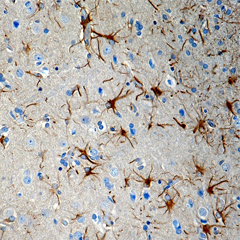
FFPE human brain tissue stained with anti-CX3CR1 antibody (clone 8E10.D9).
MojoSort™ Mouse CX3CR1 Selection Kit
This magnetic bead-based cell separation kit allows isolation or depletion of CX3CR1+ microglia and their use in downstream applications such as functional assays, gene expression and phenotypic characterization.
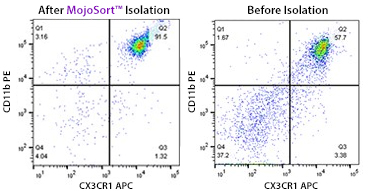
A single cell suspension from C57BL/6 mouse brain was prepared using Trypsin digestion and 70/37/30% percoll gradient to isolate CX3CR1+microglia using the MojoSort™ Mouse CX3CR1 Selection Kit. Cells were gated on CD11b and CX3CR1. The bottom plot shows cells after percoll fractionation. Debris and Dead cells were gated out with 7-AAD viability dye.
Purified anti-P2RY12 Antibody (clone S16007D)
Clone S16007D demonstrates specificity for murine P2RY12 and is effective for use in IHC, IF, and FC.
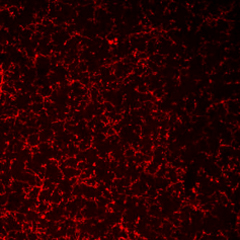
PFA-fixed frozen mouse brain tissue stained with anti-P2RY12 antibody (clone S16007D).
PE anti-human P2RY12 Antibody (clone S16001E)
Clone S16001E demonstrates specificity for human P2RY12 and is effective for use in FC.
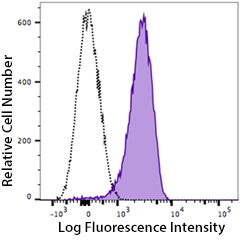
Human peripheral blood platelets stained with PE anti-P2RY12 antibody (clone S16001E, filled histogram) or PE mouse IgG2a isotype control (open histogram).
ProductsHere



Follow Us