 Login / Register
Login / Register 



Astrocytes are the most abundant glial cell type residing in the brain, accounting for 20- 40% of all glial cells. Astrocytes have many important functions, some of which include neurotransmitter uptake and release, modulation of synaptic transmission, and nervous system repair. This webpage covers a background summary of astrocytes, highlights an important astrocytic marker, and the contribution of astrocytes to Amyotrophic Lateral Sclerosis (ALS) and Autism Spectrum Disorders.
Loading...
Astrocytes
Astrocytes perform many functions. They provide metabolic support to neurons and can rapidly remove excess neurotransmitter (e.g. glutamate) released into the synaptic cleft to protect neurons against neurotoxicity. In addition, astrocytes help maintain brain blood barrier (BBB) integrity and permeability by projecting astrocytic endfeet to encircle and cover endothelial cells of the blood vessels. Similar to microglia, astrocytes are highly sensitive to alterations in their microenvironment, and undergo morphological changes and alter their gene expression profile to upregulate expression and secretion of a variety of bioactive molecules, such as cytokines and chemokines, in response to CNS injury. Furthermore, the function of astrocytes in the maintenance of the BBB has significant implications during inflammation as dysfunction of astrocytes may lead to BBB disruption and favor the infiltration of peripheral immune cells and molecules into the CNS. Astrocytes are macroglial cells in the central nervous system (CNS) and can be identified with markers such as EAAT1/Glast, EAAT2/GLT-1, ALDH1L1, glutamine synthetase, S100β, and Glial Fibrillary Acidic Protein (GFAP).
GFAP is a member of the intermediate filament (IF) family of proteins, and is specifically expressed in astrocytes. IF proteins, together with microtubules and microfilaments, form an interconnected network of the cytoskeleton, which gives cells their form, shape and functions. Astrocytes respond to damage and many disease states resulting in "astrogliosis" or the presence of a "glial response". GFAP antibodies are widely used to detect reactive astrocytes which form part of this response, since reactive astrocytes stain much more strongly with GFAP antibodies than normal astrocytes. GFAP is also a major component of the “glial scar”, an astrocyte rich structure that can inhibit nerve fiber regeneration following damage in the central nervous system. Neural stem cells frequently strongly express GFAP. Antibodies to GFAP are therefore very useful as markers of normal and reactive astrocytic cells and neural stem cells. Visit the Featured Products tab to view a few of our highly specific GFAP antibodies.

Schematic representation of a ‘tripartite synapse’, which refers to the localization of astrocytes in the proximity of synapses. Astrocytes can communicate bi-directionally to relay information to both pre- and post-synaptic structures. This communication is facilitated through the release of gliotransmitters (e.g. glutamate and ATP) from astrocytes. In this figure, glutamate released by neurons is cleared from the synapse and recycled in astrocytes to generate glutamine, which is the essential component of glutamate production at the nerve terminals.
Amyotrophic Lateral Sclerosis (ALS), commonly known as Lou Gehrig’s disease, is a neurodegenerative disorder that affects motor neurons in the motor cortex, brain stem, and spinal cord. Genetic mutations in superoxide dismutase 1 (SOD1), TAR DNA binding protein-43 (TDP-43), and fused in sarcoma (FUS) have been linked to familial forms of ALS. These mutations lead to abnormalities in cellular pathways such as RNA metabolism, mitochondrial dysfunction, and glial-mediated neuroinflammation. Astrocytes have been implicated as a contributing factor in ALS pathology and disease progression. Astrocytes in ALS lesions demonstrate altered morphology and gene expression patterns that correlate with a reactive state. Furthermore, astrocytes derived from ALS patients and a SOD1 transgenic mouse model show increased inflammatory cytokine and chemokine gene expression (e.g. CCL2, CCL11, IL-8, CXCL1).
In addition to phenotypic changes, astrocytes in SOD1-ALS models show other features, such as ubiquitinated SOD1 species reflecting defective proteostasis, elevated levels of reactive oxygen species (ROS), and secretion of TGF-β1, which prevents neuroprotective functions of microglia and T cells. Furthermore, astrocytes in the spinal cord of SOD1 mice and sporadic ALS patients, have elevated levels of the gap junction protein, connexin 43, leading to increased intracellular calcium levels and toxicity in motor neurons.

Astrocytes lose their homeostatic function in ALS and secrete cytotoxic factors detrimental to motor neurons' health.
Reference:
- Yamanaka K & Komine O. 2018. Neuroscience Research. 126:31. Pubmed
Astrocytes play an important role in the development of functional neural circuitry by providing factors that lead to proper neuronal migration, dendritic growth, and synapse formation. Recent studies have begun elucidating the involvement of astrocytes in a class of syndromes known as autism spectrum disorders (ASD). For instance, astrocytes from patients with autism show decreased branching processes, elevated GFAP expression levels in CSF, and altered brain region-specific aquaporin 4 and connexin 43 expression.
Rett syndrome, an X-linked ASD disorder, is caused by mutations in the transcription factor MeCP2. Disease progression and pathology in Rett syndrome is party attributed to loss of MeCP2 function in astrocytes and secretion of factors that are toxic to neurons. Co-culturing Mecp2−/− astrocytes with wild-type neurons has been shown to compromise neuronal integrity, and leads to the development of shorter and fewer dendrites.

Wild-type neurons produce elaborate dendrites when co-cultured with wild-type astrocytes (A) but show decreased dendritic arbors in the presence of Mecp2−/− astrocytes (B). Similarly, conditioned media from wild-type astrocytes provides neurotrophic support for neurons (C), in contrast to conditioned media from Mecp2−/−astrocytes (D), which is toxic to neurons.
BioLegend offers the largest collection of GFAP-specific Sternberger (SMI®) monoclonal antibodies (SMI 21-26) that are the gold-standard for visualization of this protein by immunohistochemical studies. These antibodies are available in purified and ascites formats, and are useful in ICC and WB in addition to IHC. Anti-GFAP SMI® clones are effective, in varying degrees, in detection of astrocytes and astrocytic processes, Bergman glia, and astrocytomas in a wide variety of species.


FFPE human brain tissue stained with
anti-GFAP antibody (clone SMI 21).

FFPE rat brain tissue stained with
anti-GFAP antibody (clone SMI 23).

FFPE mouse brain tissue stained with
anti-GFAP antibody (clone SMI 24).

FFPE rat brain tissue stained with
anti-GFAP antibody (clone SMI 23).
Purified anti-GFAP Antibody (clone SMI 24)
Antibodies against GFAP are used as a cell-specific marker to distinguish astrocytes from other glial cells in the CNS. Clone SMI 24 is effective for use in IHC and WB
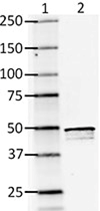
Western blot of clone SMI 24
Ln 1= Molecular weight marker
Ln 2= Rat brain lysate
Purified anti-S100B Antibody (clone 11C12E12)
Clone 11C12E12 is used for detection of astrocytes in human, mouse and rat brain tissues. This clone is effective for use in WB.
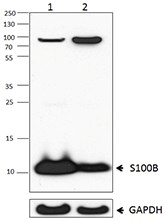
Western blot of clone 11C12E12
Ln 1= Rat brain lysate
Ln 2= Mouse brain lysate
Alexa Fluor® 647 anti-GFAP Antibody (clone 2E1.E9)
Clone 2E1.E9 is used for detection of astrocytes in human, mouse and rat brain tissues. This clone is effective for use in IF and IHC.

FFPE human cerebellum tissue stained with Alexa Fluor® 647 anti-GFAP (clone 2E1.E9, red) and Alexa Fluor® 594 anti-Tubulin Beta 3 (TUBB3) (clone AA10, magenta) antibodies. Nuclei were counterstained with DAPI (green).
ProductsHere



Follow Us