Autofluorescence
Different cell types and tissues, as well as media additives, have varying levels of inherent fluorescence. Major sources of autofluorescence (AF) include NADH, riboflavins, flavin coenzyme molecules, crosslinking of primary amines by paraformaldehyde, and certain biological structures (e.g., mitochondria, lysosomes). Myeloid cell lineages tend to be particularly problematic with AF. These proteins and molecules are more easily excited at lower wavelengths (i.e. from the UV, violet, and blue lasers) and will emit at a wide range of 500-700 nm, which overlaps several common fluors like FITC and PE. You should always include a unstained control to check for the level of AF.
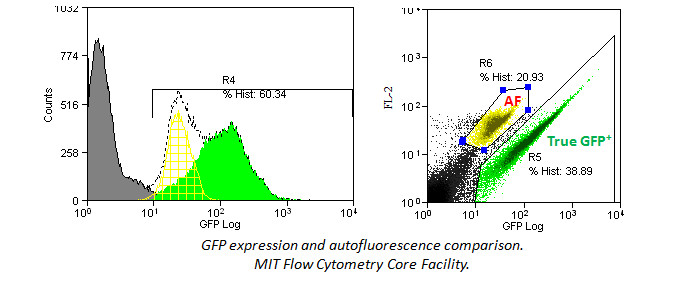
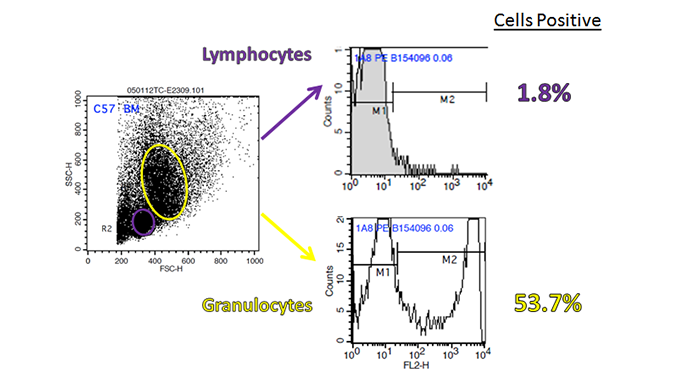
In addition, histograms may hide an autoflorescent population. In the example below, a bivariate comparison of FL-1 (GFP) and FL-2 for a GFP-expressing cell line reveals an autofluorescent population that can be gated out.
Dilution
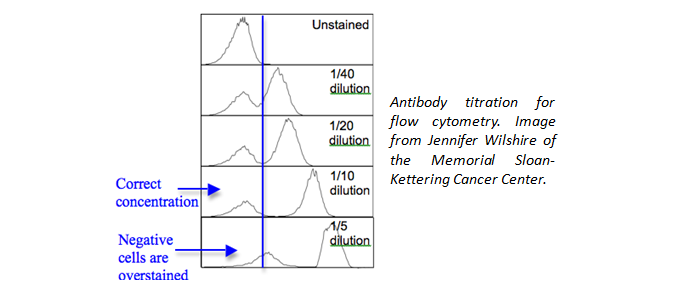
Is the proper amount of antibody being used? Although we have recommended amounts to be used, you could also titrate the antibody down to find an optimal amount to use for your unique experiment. This will help to eliminate background staining and improve your signal to noise ratio.
High Compensation
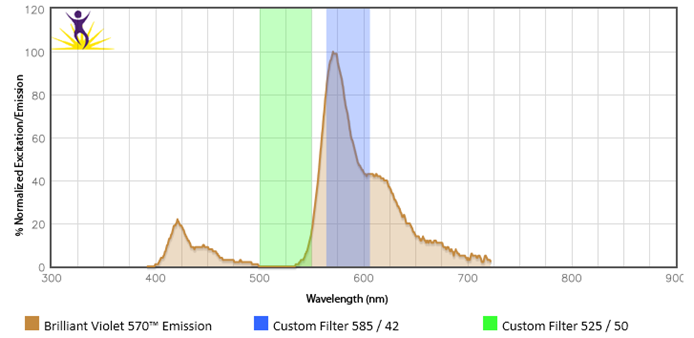
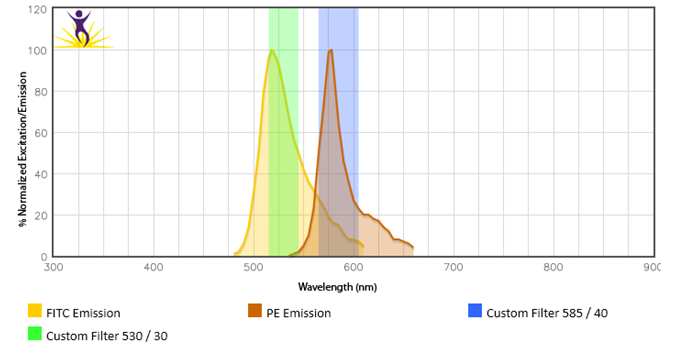
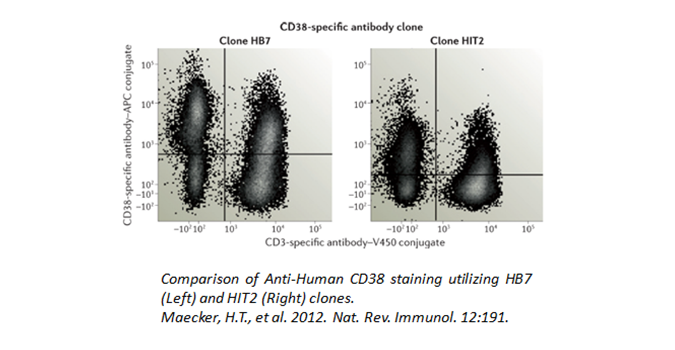
When two or more fluorophores overlap in emission spectra, some compensation will need to be applied. Depending on the amount of spillover, potentially high levels of compensation may be needed. In these cases, voltages that are being used for each PMT need to be balanced, because excessive differences in the voltage can amplify the requirement for compensation. For instance, BV510™ and BV570™ are known to require compensation because BV510™ will spillover into the BV570™ channel and vice versa. If the BV570™ channel’s voltage is much higher than the BV510™ voltage, this will amplify the spillover. Notice in the example below, when the PMTs are balanced the requirement for compensation is greatly reduced. If the appropriate levels of compensation are not applied, your background may appear artificially high.

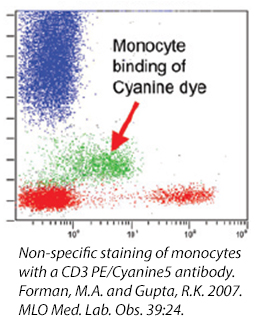
Cyanine Dyes
Cyanine dyes and some of their tandems have a high propensity for binding to Fc Receptors, particularly monocytes. This reaction cannot be prevented with the use of a Fc Blocking reagent and its exact cause is unknown. If you are studying a population particularly high in myeloid-type cells (i.e., bone marrow) , you may want to consider using other fluors to denote your populations. Or, take a look at our True-Stain Monocyte Blocker™, which is specifically desgined to stop this interaction.
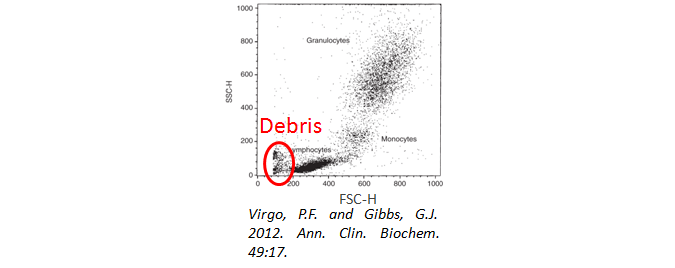
Cell Debris
Have you used a viability dye to ensure your cells are alive and happy during analysis? Cell debris and dead cells can lead to high background staining. In addition, when gating your cells, be sure to exclude the debris from your analysis. They will often have a very low forward and side scatter, as seen below.
Viability
If dead cells/debris were to be accidentally included in your analysis, you may see false positive populations. Staining with Propidium Iodide or 7-AAD, which only enter into dead cells, is an excellent method to identify and exclude dead cells from your analysis.
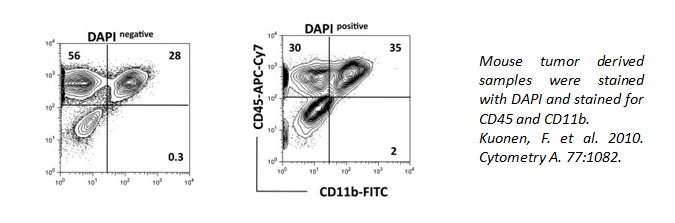
DAPI can also be used to identify live/dead cells. It can also pass through live cell membranes, but in a less efficient manner. Below, the staining of CD11b and CD45 is altered within the DAPI+ population of cells compared to DAPI- cells.
For samples that need to be fixed and permeabilized (particularly with ethanol or methanol), DNA intercalating dyes like propidium iodide and 7-AAD are often not suitable. This is because the DNA can be unwound, popping the dyes off and allowing them to non-specifically bind cells that are now permeabilized. For these experiments, we would highly recommend Zombie Dyes, which bind to primary amines instead of DNA.
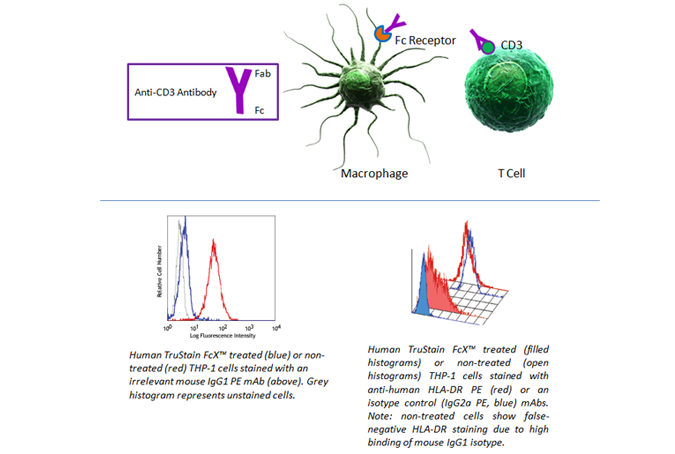
Fc Blocking
Fc receptors are present on many cell types, including granulocytes, B cells, macrophages, and dendritic cells. As they have a propensity to bind the Fc portion of antibodies, they can present false positives in your analysis. To prevent this background staining, we recommend using Human TruStain FcX™ (Fc Receptor Blocking Solution) (Cat. No. 422301) or TruStain FcX™ PLUS (anti-mouse CD16/32) Antibody (Cat. No. 156604).
FMOs/Isotype Controls
Isotype controls are negative controls designed to help indicate the non-specific background staining (due to Fc Receptor or cellular protein interaction) of your test antibody. FMOs, or Fluorescence Minus One controls, can also be helpful, particularly if your antigen of interest is lowly expressed or you have a complex multicolor (4 or more) panel. This will help you observe any spillover from the other fluors in your panel and aid your selection of a true positive population.
You should also make sure to use the same amount of test and isotype control antibody. If you used 1 ug of Anti-CD4, use 1 ug of its corresponding isotype control.
Avoid purchasing isotypes and test antibodies from different companies, as the fluorophore:protein (F:P) ratio may not be consistent between different manufacturers.

Doublets
Doublets can occur when cells clump together or are in the process of dividing. This presents difficulties when analyzing flow cytometry, as the cell mass will still count as one event. As such, if you do not gate them out of your analysis, they can lead you to misleading data. While it may seem like a double positive population may be present in your analysis, you should make sure it is not the result of two merged cells, with each cell expressing only a single marker.
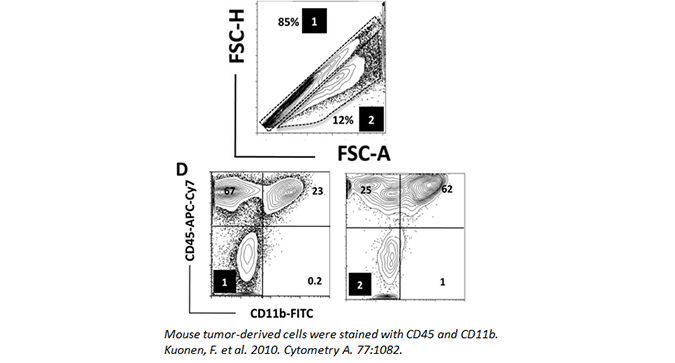
Doublets can be minimized by adding EDTA to your FACS buffer and filtering your samples through a 30 micron filter. Doublets can also be easily gated out by assessing cell width, height, and area (as seen below). In the data, (1) denotes the singlet population, while (2) marks the doublets in the sample. The inclusion of doublets in your analysis can skew your results.
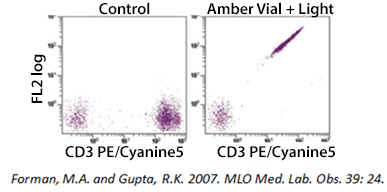
Tandem Degradation
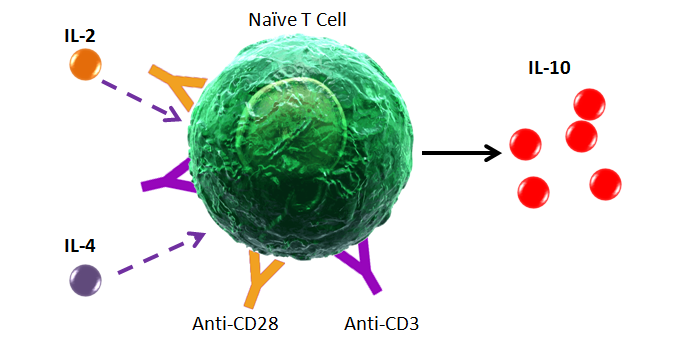
If you are using a tandem dye, breakdown can occur if the antibody is not stored optimally and protected from light exposure. In the example below, a PE-Cyanine5 dye has been exposed to light. As the tandem breaks apart, a signal can be detected in the FL2 (PE) channel. Tandem dyes are also more sensitive to certain fixation methods. We recommend FluoroFix™ Buffer for fixation of samples using tandem dyes. You can learn more about tandem dyes from the link below:
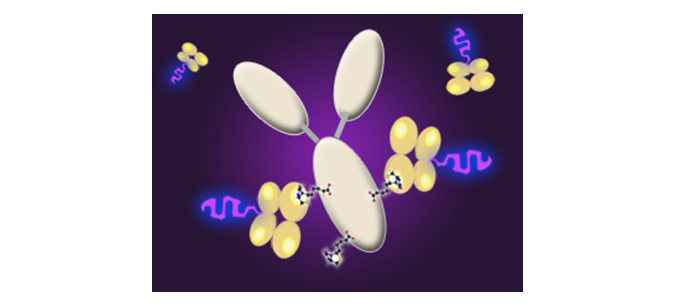
Tandem Dyes
 Login / Register
Login / Register 


















Follow Us